
Лечение вегето-сосудистой дистонии у детей и подростков
Вегетативная дисфункция нервной системы является частой проблемой детей в подростковом возрасте. Это связано с гормональной перестройкой организма, бурным ростом и наследственными факторами. Мощный выброс половых гормонов приводит к нестабильности сердечно-сосудистой и нервной систем. На фоне гормональной перестройки у детей часто повышается эмоциональный фон, появляется нестабильность настроения, что дополнительно негативно влияет на функцию сердечно-сосудистой системы и приводит к повышению или понижению артериального давления.
Вегето-сосудистая дистония: симптомы. Диагноз ВСД
Бурный рост организма может способствовать возникновению дефицита кровотока в шейном отделе позвоночника ввиду ослабления компенсаторных механизмов организма, что приводит к появлению головокружения при резкой перемене положения тела. Все выше сказанное может вызывать склонность к повышению или понижению артериального давления, нарушение сердечного ритма, обмороки, головокружения.
Обследование и лечение детей с вегетативными расстройствами направлено на выявление нарушений и улучшение функций сердечно-сосудистой, эндокринной и нервной систем, компенсацию внутренних резервов организма, стабилизацию эмоционального фона и носит комплексный характер.
ВСД: лечение
В комплекс лечения детей с вегетативной дисфункцией входит:
- Терапия лекарственными препаратами.
- Физиолечение.
- Галотерапия.
- Массаж воротниковой зоны или волосистой части головы.
В зависимости от степени выраженности вегетативных нарушений лекарственные средства назначаются внутрь, внутримышечно или внутривенно капельно. При наличии у ребенка частых головных болей, нестабильности артериального давления (с частым повышением цифр более 150 мм.рт.ст.) возможен вариант лечения в условиях дневного стационара под наблюдением врача-невролога и медицинской сестры с мониторингом состояния здоровья в течение 7-10 дней.
В комплекс лечения детей с вегетативной дисфункцией входит:
- Формирование полноценного сна не менее 8-9 часов с периодом засыпания не позднее 23 часов.
- Ведение активного образа жизни с минимизацией длительных статических поз — занятия спортом, фитнес, плавание, ходьба пешком.
- Чередование умственного и физического труда
- Дозированная зрительная нагрузка, исключить злоупотребление гаджетами.
При соблюдении всех рекомендаций по лечению состояние здоровья пациентов стабилизируется, прогноз благоприятен.
Записаться к неврологу
РадикулопатияРадикулопатия — это ущемление корешков спинного мозга с последующим развитием воспалительных процессов…
Задержка речевого развития у детей (ЗРР)Задержка речевого развития (ЗРР) является частой причиной обращения родителей к неврологу. Темпы развития…
Бессонница у детейБессонница у ребёнка: что делать? Здоровый сон очень важен для малышей. Продолжительность ночного отдыха…
В настоящее время синдром дефицита внимания с гиперактивностью (СДВГ) широко распространен в детской…
ЭнурезЭнурез – недержание мочи, которому подвержены дети в возрасте от 5 лет. Современные врачи считают патологическим…
КинезиотейпированиеКинезиотейпирование — один из методов, применяемый в восстановительной медицине и реабилитации. Дословно…
Первый детский медицинский центр
Здоровье детей – спокойствие родителей!
симптомы и лечение у подростков
Еще 20 лет назад о вегето-сосудистой дистонии у подростков мало кто знал. Сейчас же заболевание набирает «популярность» вреди этой возрастной категории детей. С чем это связано, каковы причины и признаки вегето-сосудистой дистонии у подростков, попробуем разобраться вместе. А так же выясним, чем опасно это состояние, каковы его последствия и какие существуют методы, чтобы избавиться от него навсегда. Обо всем об этом мы и поговорим в этой статье.
Сейчас же заболевание набирает «популярность» вреди этой возрастной категории детей. С чем это связано, каковы причины и признаки вегето-сосудистой дистонии у подростков, попробуем разобраться вместе. А так же выясним, чем опасно это состояние, каковы его последствия и какие существуют методы, чтобы избавиться от него навсегда. Обо всем об этом мы и поговорим в этой статье.
Что такое вегето-сосудистая дистония
Вегето-сосудистая дистония, к сожалению, очень распространена среди подростков на сегодняшний день. Практически 9 из 10 подростков получают такой диагноз после посещения доктора. Но понять, что «болит у организма» довольно сложно, ведь внешне все выглядит как банальное недомогание.
Вегето-сосудистая дистония – это совокупность нескольких симптомов. ВСД (так в медицине сокращенно называют это заболевание) заключается в проявлении необычной реакции на стресс или смену привычных условий. Многие люди считают такую реакцию вполне нормальной. Чаще всего вегето-сосудистая дистония сопровождается сердечными болями, нехваткой воздуха для дыхания, раздражительностью или повышенной утомляемостью.
В подростковом возрасте ВСД может протекать как в острой, так и в хронической форме и чаще всего возникает на фоне устойчивых стрессовых перенапряжений. Наиболее распространено это состояние в 12-15летнем возрасте. ВСД не болезнь, а общее состояние организма, вызванное комплексом общих проблем, связанных с нарушением крообращения и кровоснабжения внутренних органов.
Причины вегето-сосудистой дистонии
Вегето-сосудистая дистония никогда не возникает спонтанно, а является следствием длительного влияния на организм негативных факторов. Неправильный образ жизни, вредные привычки как раз и становятся основными причинами развития такого состояния. Основными причинами вегето-сосудистой дистонии у подростков являются:
- Наличие вредных привычек;
- Гормональная перестройка;
- Недавно перенесенные тяжелые заболевания;
- Малоподвижный образ жизни;
- Стрессы, связанные с взаимоотношениями;
- Чрезмерная нагрузка на учебе.

Для большинства подростков характерны эмоциональные нагрузки, из-за которых и возникает проблема вегето-сосудистой дистонии. В связи с изменение психики, подростки становятся более уязвимыми и легче подвергаются влиянию со стороны. Так, конфликты в школе или семье, чрезмерное употребление алкоголя или табака, загруженность на учебе или проведение большого количества времени в неподвижном состоянии, например, за компьютером, могут неблагоприятно повлиять на еще не окрепшую нервную систему подростка.
Главным пусковым механизмом возникновения вегето-сосудистой дистонии у детей подросткового возраста является нарушение роста периферической нервной системы, а именно ее замедление на фоне роста мышечной ткани.
Причины появления ВСД у юношей обычно таковы: быстрое половое созревание приводит к чрезмерным силовым нагрузкам, оказывающим чрезмерную нагрузку на сердце. Также юноши раньше начинают проявлять интерес к алкоголю, наркотикам и сигаретам, что тоже влияет на развитие вегето-сосудистой дистонии.
Девушки очень эмоциональны, а потому ВСД появляется от долгого обдумывания неприятной для девушки темы, чрезмерные переживания по поводу оценок окружающих, общественного мнения, ссоры с подругой или парнем. Во время менструации девушки также подвержены нервным расстройствам, связанным с вегето-сосудистой дистонией.
Наконец, вегето-сосудистая дистония может передаваться по наследству: нарушения сердечно-сосудистой системы у родителей обязательно отобразятся на здоровье их ребенка. Поэтому, если в семье есть предрасположенность к сердечным заболеваниям, то родители должны более тщательно следить за здоровьем своего ребенка, закаливать его с раннего детства, развивать физически.
Признаки вегето-сосудистой дистонии
Признаки вегето-сосудистой дистонии у подростков могут быть абсолютно разными. Чаще всего это быстрая утомляемость, чрезмерная раздражительность, потливость, бессонница ночью и сонливость днем, головные боли и затруднение дыхания.
Что касается настроения подростка во время ВСД, то появляется чрезмерная плаксивость и подавленность. Нередко у подростков появляются различные страхи и панические атаки, наблюдается истеричность подростков и депрессивность.
Вегето-сосудистая дистония, в зависимости от организма подростка, сопровождается либо повышением аппетита, либо его понижением. Боли в животе, тошнота и нарушение выделительной системы также являются симптомами ВСД.
Из-за вегето-сосудистой дистонии кожа подростка приобретает мраморный оттенок и, в отдельных случаях, отекает. Руки и ноги постоянно холодные, даже если на улице или в помещении достаточно тепло.
Диагностика вегето-сосудистой дистонии
Диагностика вегето-сосудистой дистонии у подростков достаточно сложна. Признаки дистонии очень непостоянны и могут быть абсолютно разными у разных людей. Определенных методов и медицинских приборов, которые способны определить вероятность наличия вегето-сосудистой дистонии у человека, в современных клиниках не существует, а потому, по неопытности или какой-либо другой причине, доктора обычно не используют этот диагноз в повседневной жизни. Многие врачи чаще всего объясняют тревожность, головокружение и перепады давления наступлением переходного возраста подростка или наследственной предрасположенностью.
Для того чтобы правильно поставить диагноз, необходимо провести множество лабораторных и инструментальных обследований и проконсультироваться с докторами разных специальностей.
Для большей точности необходимо проверить наличие вегето-сосудистой дистонии у родственников подростка, а также пройти обследование у ЛОРа, невролога, психотерапевта, эндокринолога, окулиста, кардиолога и педиатра. Только после всего вышеописанного диагноз, поставленный в клинике, будет наиболее точным и правдивым.
Только после всего вышеописанного диагноз, поставленный в клинике, будет наиболее точным и правдивым.
Лечение болезни у подростков
Лечение ВСД у детей подросткового возраста подразумевает комплексный подход к проблеме. Часто задействуется несколько узкопрофильных специалистов, пытающихся помочь справиться ребенку с проблемой. Своевременная терапия дает благоприятный прогноз, позволяющий обойтись не медикаментозными методами.
В целом же лечение вегето-сосудистой дистонии индивидуально и делится на два типа:
- Медикаментозное;
- Не медикаментозное.
Медикаментозное лечение применяется в особо тяжелых случаях вегето-сосудистой дистонии у подростков. В этом случае подросток нуждается в восстановлении нервной системы. Ему прописываются такие ноотропы, как Пинтогам, Пирацетам или Кавинтон. Также необходимо применение подростком витаминов и минералов. Особенная нехватка в организме при ВСД витамина B.
Различные психосоматические реакции требуют различных методов борьбы с ними. Так, в зависимости от степени выраженности подростку прописываются антидепрессанты, транквилизаторы или нейролептики. Но использование этих средств рекомендовано только с назначения психоневролога.
Также при медикаментозном лечении нередко используются различные травяные отвары. Чаще всего используются женьшень, солодка и золотой корень, а также лимонник и заманиха.
При медикаментозном лечении есть противопоказания, поэтому категорически запрещено использовать лекарственные средства без назначения врача-специалиста. Также запрещено длительное лечение медикаментами, чтобы у подростка не возникло привыкания.
Не медикаментозное лечение представляет собой ряд не медикаментозных рекомендаций, таких как:
- Длительный сон и прогулки на свежем воздухе;
- Сокращение времени пользования компьютером и телевизором;
- Занятия подвижным видом спорта;
- Массаж;
- Прослушивание успокаивающей музыки;
- Правильное питание;
- Фиточаи;
- Психотерапия;
- Иглорефлексотерапия.

Кроме этого рекомендуются и специальные методы лечебной гимнастики и релаксации. Курс лечения и подбор упражнений обсуждается в лечащим врачом в индивидуальном порядке.
Последствия вегето-сосудистой дистонии
Обычно последствия вегето-сосудистой дистонии не очень серьезны. Большинство подростков избавляется от нее еще в подростковом возрасте и только 15% продолжают страдать от данной болезни. У многих людей, имевших диагноз ВСД в юности, к 30-ти годам возможно частое повышение давления и появление гипертонии.
В том случае, если в нужное время подростку не оказали помощь в лечении вегето-сосудистой дистонии, в будущем эта болезнь будет только развиваться и приобретать более тяжелые симптомы, а значит и избавиться от болезни будет значительно труднее, чем в подростковом возрасте.
При отсутствии лечения или при неправильной терапии во взрослом возрасте у человека могут появиться различные страхи и фобии, избавиться от которых поможет только высококвалифицированный специалист. Лечение у психотерапевта необходимо и тем пациентам, у которых пусковым механизмом к развитию ВСД стали хроническая усталость, недосыпание, устойчивые стрессовые перенапряжения, другие депрессивные состояния.
Профилактика вегето-сосудистой дистонии
Для того чтобы избежать развития вегето-сосудистой дистонии у подростков необходимо соблюдать несколько простых профилактических правил:
- Соблюдение режима дня;
- Сбалансированное, правильное питание;
- Наличие полезных привычек;
- Отказ от вредных привычек;
- Занятия подвижным видом спорта;
- Закаливание.
Также необходимо создать подростку все условия для спокойного образа жизни, который не сможет нарушить нервную систему. При наличии родственников, имеющих вегето-сосудистую дистонию, подросток должен находиться под наблюдением невропатолога.
Немаловажную роль играет способность к саморегулированию и самовнушению. Такие подростки склонны к спокойствию и могут легко овладеть любой ситуацией.
Соблюдение режима дня очень влияет на самочувствие человека и его нервную систему, поэтому, во избежание вегето-сосудистой дистонии, необходимо уделять 8 часов сну, 2 часа прогулкам на свежем воздухе и не более часа использованию компьютеров и телевизоров. А правильное питание подразумевает сокращение потребления мясных продуктов, кофе и крепких чаев, выпечки и сладостей и включение в повседневный рацион свежие фрукты и овощи, крупы и не менее 1,5 литра чистой воды в сутки.
Высококвалифицированные специалисты уверены, что вегето-сосудистая дистония – это лучшее, что может случиться с подростком, а потому переживать по этому поводу родителям не стоит. Эта проблема обязательно пройдет с возрастом, и подросток вновь станет здоровым и активным. Единственная возможная опасность при вегето-сосудистой дистонии – это повышенное давление, которое вызывает другие заболевания, связанные с сердцем. Не стоит пренебрегать профилактикой данной болезни, обязательно рекомендовано постоянное соблюдение режима дня, правильное питание и занятия спортом. Все это поможет подростку не только избежать вегето-сосудистой дистонии, но и предотвратить многие другие опасные заболевания.
Вместо заключения
Чтобы избежать в будущем таких серьезных патологий со здоровьем, следите за привычками своего ребенка с раннего возраста. Приучайте его к спорту, походам, активному образу жизни. Поменьше телевизора и планшета, побольше прогулок и положительных эмоций и вегето-сосудистая дистония некогда не постучится к вам в дом.Вегето-сосудистая дистония в подростковом возрасте — Управління охорони здоров’я м. Миколаєва
Вегето-сосудистая дистония, по разным оценкам, встречается у 20- 45% подростков. Такой большой разброс связан с тем, что ВСД — не болезнь, а синдром, совокупность симптомов. Это нарушение нормальной реакции организма на стресс, нагрузки, смену привычных условий. И нередко сам человек и окружающие могут считать такую реакцию нормой. Проявления вегето-сосудистой дистонии могут быть самые разные: сердечные (боль, учащенное сердцебиение, ощущение сбоев ритма сердца), дыхательные (чувство нехватки воздуха), колебания артериального давления, расстройства работы желудочно-кишечного тракта, повышенная утомляемость, раздражительность и т. д.
Проявления вегето-сосудистой дистонии могут быть самые разные: сердечные (боль, учащенное сердцебиение, ощущение сбоев ритма сердца), дыхательные (чувство нехватки воздуха), колебания артериального давления, расстройства работы желудочно-кишечного тракта, повышенная утомляемость, раздражительность и т. д.
Подростки на острие главного удара ВСД. Именно у подростков вегето-сосудистая дистония возникает особенно часто и именно у этой возрастной группы она является наиболее серьезной проблемой.
Во-первых, одна из самых распространенных причин ее развития — рассогласование физического развития и зрелости нервной системы. Да и гормональная перестройка организма, происходящая в пубертатном периоде, может стать провоцирующим фактором.
Во-вторых, сама жизнь современного подростка — прекрасный повод для развития вегето-сосудистой дистонии. С одной стороны, огромные учебные нагрузки, стрессы, связанные с взаимоотношениями со сверстниками и родителями. С другой — недостаточная физическая активность, которая у детей сегодня отнюдь не редкость, нарушения режима сна/бодрствования из-за «ночного зависания» в интернете, бесконечных компьютерных игр и т. д. При этом вегето-сосудистая дистония сама по себе может сильно осложнить жизнь ребенка, превращая ее в порочный круг: учебные нагрузки провоцируют стрессы, стрессы приводят к симптомам ВСД, которые мешают учиться, а проблемы с учебой, в свою очередь, приводят к новым стрессам.
Воспаление хитрости?
Ряд проблем связан с тем, что родители не всегда правильно реагируют на то, что у их ребенка вегето-сосудистая дистония. Одни очень долго не обращают внимания на проявления синдрома, считая их капризами, притворством — и доводят до того, что у ребенка начинаются настоящие кризы. Другие, напротив, едва услышав словосочетание «вегето-сосудистая дистония» (а иногда и поставив этот диагноз самостоятельно, без помощи врача), начинают обращаться с ребенком как с больным: постоянно измеряют ему температуру и артериальное давление, пичкают лекарствами и народными средствами, запрещают любую физическую нагрузку, от физкультуры в школе до походов и подвижных игр. Наконец, вегето-сосудистая дистония у подростка, как правило, прогрессирует с возрастом, если ее не лечить.
Наконец, вегето-сосудистая дистония у подростка, как правило, прогрессирует с возрастом, если ее не лечить.
Как лечить ВСД?
Лечение вегето-сосудистой дистонии должно быть очень индивидуальным и зависит оттого, как она проявляется. В первую очередь необходимо нормализовать режим дня: спать не меньше 8 часов в сутки, ложиться и вставать в определенное время, не пропускать приемы пищи, хотя бы час в день находиться на свежем воздухе, поменьше смотреть телевизор. Все эти прописные истины, как ни странно, — мощные средства профилактики развития ВСД.
Следует уделить внимание и питанию. Лишний вес, избыточное потребление сладкого, мучного, жиров — также факторы развития синдрома. А вот, например, крепкий чай или хороший молотый кофе могут оказаться полезными для здоровья ребенка — если ВСД проявляется в виде сниженного артериального давления, вялости. Обогатите пищу подростка продуктами, богатыми калием: курагой, орехами, зеленью, фасолью.
Купировать постоянные проявления ВСД способны и лекарства, но назначить их должен врач. Правильно подобранные препараты не только устранят симптомы тревоги и страхи, но и помогут избавиться от аритмии, повышенного артериального давления, болевых ощущений. Однако отдавать предпочтение все же стоит немедикаментозным средствам.
Л.А.Кузьмина, врач–педиатр
отделения «Клиника, дружественная к молодежи»
ГДП №4.
Диагностика вегето-сосудистой дистонии, ее симптомы и лечение
Вегето-сосудистая дистония или нейроциркулярная дисфункция представляет собой нарушение нервной системы, характеризующееся общим ухудшением самочувствия человека. Чаще всего вегето-сосудистая дистония диагностируется у детей или подростков и вызывает проблемы с функционированием всего организма. Наиболее часто этот недуг обнаруживают у девочек в возрасте 7-8 лет.
Симптомы
Ввиду того, что ВСД влияет на весь организм, симптомы также могут проявляться в одной или нескольких его системах. Ниже перечислены наиболее распространенные симптомы, встречающиеся при нейроциркулярной дисфункции, в зависимости от типа заболевания: симпатикотония или ваготония.
Ниже перечислены наиболее распространенные симптомы, встречающиеся при нейроциркулярной дисфункции, в зависимости от типа заболевания: симпатикотония или ваготония.
Общие нарушения состояния организма:
- высокая утомляемость;
- слабость;
- проблемы со сном в виде бессонницы или сонливости;
- плохое настроение.
Нарушения по системам.
Нервная система:
Пищеварение:
- проблемы с кишечником;
- боли в животе.
Сердечнососудистая система::
- боли и шумы в сердце;
- изменение артериального давления.
Дыхательная система:
- затрудненное дыхание;
- частая одышка, даже при небольшой нагрузке.
Мочевыделительная система:
- увеличение частоты мочеиспускания;
- учащение позывов в ночное время.
Обычно болезнь протекает в вялой форме или же беспокоит пациента приступами, их также называют вегетативные кризы. Кризы длятся от нескольких минут, до нескольких часов. Эмоциональные переживания, физическое или умственное напряжение, инфекции – все это может стать катализатором очередного приступа.
Диагностика
Диагностика ВСД в домашних условиях невозможна, ввиду того, что для обнаружения болезни необходимо обследование всех органов, беспокоящих пациента. Для диагностирования проводятся следующие манипуляции:
- расшифровка анализов мочи и крови;
- электрокардиография;
- эхокардиография;
- дыхательные тесты;
- УЗИ органов брюшной полости;
- электроэнцефалография;
- эхоэнцефалография.
Также при выявлении заболевания важно изучить наследственную информацию больного.
Врачи отмечают, что при симпатикотонии у больных обнаруживались наследственные болезни, такие как, сахарный диабет или гипертоническая болезнь, а при ваготонии – нейродермит, язва или астма. Именно генетическая предрасположенность является основным фактором, влияющим на риск развития ВСД.
Именно генетическая предрасположенность является основным фактором, влияющим на риск развития ВСД.
Лечение
Несмотря на генетическую предрасположенность к заболеванию, его развитие можно предотвратить, при условии соблюдения режима, дозированного времени для работы и отдыха, а также правильного питания. Так, если у пациента отмечается повышенное артериальное давление – стоит уменьшить количество потребляемой соли, кондитерских изделий, жирных продуктов, а также жареной пищи. При пониженном давлении врачи рекомендуют не увлекаться продуктами, содержащими йод, натрий и калий в больших количествах, соблюдать режим сна, не переутомляться, избегать стрессовых состояний.
В борьбе с недугом специалисты делают упор на немедикаментозный метод, а именно:
Для снижения и облегчения симптоматики используют лекарственные травы:
- элеутерококк;
- корень женьшеня;
- заманиха;
- аралия;
- пустырник;
- валериана;
- боярышник.
В случае низкой эффективности описанных методов доктора прибегают к использованию фармацевтических препаратов, которые подбираются отдельно для каждого пациента. Кроме того могут быть использованы антидепрессанты, успокоительные средства, витаминно-минеральные комплексы, препараты восстанавливающие кровоснабжение мозга.
Для успешного оздоровления требуется регулярное обследование – раз в 3-6 месяцев. Нейроциркулярная дисфункция, как правило, усиливается весной и осенью, поэтому все лечебные мероприятия лучше назначать именно на это время года.
В случае несвоевременного или некачественного лечения дистония прогрессирует и приводит к появлению самых разных патологий в организме, если же соблюдать предписания врача и следить за течением болезни – прогнозы благоприятные.
К какому врачу обратиться
Наблюдением и лечением больных ВСД занимается невролог. Также в избавлении от недуга принимают участие физиотерапевт, специалист по лечебной физкультуре, массажист, рефлексотерапевт.
Для диагностики также потребуется консультация следующих специалистов:
Вегето-сосудистая дистония (ВСД, НЦД) – Корсаков
Как ни странно, один из самых распространенных «легких» терапевтических, кардиологических и неврологических диагнозов – вегето-сосудистая дистония/ВСД/НЦД – нейроциркуляторная дистония, просто отсутствует в классификации МКБ-Х. В итоге многим врачам, проще всего выставить несуществующий диагноз и назначить неэффективное лечение, чем вести кропотливый диагностический поиск.
Нигде в мире, за исключением постсоветского пространства данный диагноз вегето-сосудистой дистонии (ВСД), а также близкие ему – нейроциркуляторные дистонии (НЦД), кардионеврозы и прочие «малые кардиологические» и «минимальные неврологические» расстройства не выставляются. Единственным вариантом, отраженным в международной классификации болезней, является – «соматоформная вегетативная дисфункция нервной системы – СВДНС», которая входит в рубрику расстройств именно психической сферы и кодируется индивидуальным шифром F.45.3.
Исходя из международных отраслевых рекомендаций лечением этого расстройства должны заниматься именно психиатры.
Проявления заболевания
Ведущими проявлениями в клинике СВДНС являются симптомы, сочетание которых уже должно насторожить терапевта или семейного врача в плане консультативной помощи психиатра. Наиболее выраженными и частыми являются следующие:
- Проявления общего характера к которым относятся повышенная утомляемость, чувство сердцебиения, общая потливость, преходящий тремор конечностей, беспричинное повышение температуры до небольших значений, общее беспокойство по поводу состояния здоровья.
- Со стороны сердечно сосудистой системы нестабильность артериального давления, чувство перебоев в работе сердца, особенно в стрессовых ситуациях, возможен незначительный дискомфорт в левой половине грудной клетки, ощущение «тяжести в сердце».
- Со стороны нервной системы головокружение и головные боли, повышенная утомляемость, метеочувствительность, легкая апатия, тяжесть в голове, онемение конечностей.
- Со стороны дыхательной системы чувство нехватки воздуха, одышка, затрудненное дыхание, форсированное дыхание.
- Со стороны пищеварительной системы изжога, тяжесть в желудке, метеоризм, аэрофагия, нарушения стула, метеоризм, редкие невыраженные разлитые боли в животе.
- Со стороны мочевыделительной системы затруднение мочеиспускания, частое обильное или, наоборот, скудное мочеиспускание.
- Невыраженные слабые летучие боли в различных суставах, а также минимальные проявления со стороны мышечной системы в виде напряжения мышц, редких судорог в отдельных группах мышц.

Важный момент
Очень важным моментом становится то, что все эти проявления сопровождаются значительным психологическим дискомфортом и общим беспокойством пациента за состояние собственного здоровья. Это в свою очередь направляет их к активному обращению к врачам, но тщательное обследование не дает результатов относительно причин возникновения указанных жалоб. В итоге, к огромному сожалению, выставляется диагноз «системного невроза» или «малых дисфункций» и назначается неэффективное лечение.
Верная тактика
Исходя из современных международных рекомендаций, обязательным шагом в таком случае становится консультирование у психиатра. В государственной системе психиатрической помощи для таких пациентов мало шансов получить качественную помощь, как минимум из-за большой загруженности врачей более серьезными «психиатрическими пациентами». Именно поэтому необходимо обращаться к частной психиатрии, где пациенту будет оказано необходимое по объему внимание, будут изучены результаты ранних обследований и назначены дополнительные уточняющие методы. После исключения любых объективных причин, возможно будет выставлен диагноз «соматоформной вегетативной дисфункции нервной системы», что соответствует общепризнанным действительным рекомендациям и будет назначено соответствующее коррекционное лечение.
Наиболее обоснованными мероприятиями становится нормализация образа жизни и привычек, избегание стрессовых ситуаций, общеукрепляющая терапия, адекватная симптоматическая коррекция, а также в некоторых ситуациях бережная седативная и анксиолитическая терапия, а также антидепрессанты. Важным моментом в лечении становится психотерапевтическая работа с пациентом.
Важным моментом в лечении становится психотерапевтическая работа с пациентом.
Вегетососудистая дистония — ГБУЗ «Кореновская ЦРБ» МЗ КК
Вегетососудистая дистония – один из самых загадочных синдромов. Большинство врачей не считают ее тяжелой болезнью, а скорее совокупностью различных болезненных проявлений. На самом деле, вегето-сосудистая дистония способна разрушить жизнь человека, существенно снизить качество жизни и даже привести к потере трудоспособности.
Вегетососудистая дистония (ВСД) — функциональное нарушение нервной системы, характеризуется нарушением общего состояния и самочувствия, проявляется неорганическими сбоями в работе различных органов и систем. Во многих медицинских изданиях можно встретить другие названия этого заболевания, а именно: вегетативная дисфункция, нейроциркуляторная дистония, невроз сердца, функциональная кардиопатия, паническая атака, ангионевроз, вазомоторная дистония и др. Сейчас все чаще встречается термин «вегетативная дисфункция» (ВД) или «вегетососудистая дистония» (ВСД).
Причины возникновения ВСД
Среди основных причин возникновения синдрома вегетососудистой дистонии врачи называют факторы наследственной предрасположенности. К примеру, чаще всего вегетососудистая дистония у детей выявляется именно как следствие наследственности. Повышенная нервозность и стрессы уже в первые месяцы беременности могут оказать значительное влияние не только на формирование личности ребенка, но и на высшую нервную деятельность головного мозга. Факты показывают, что эмоциональная неустойчивость детского организма провоцирует развитие ВСД даже в детские годы. Подростковые годы являются переходными не только в процессе превращения ребенка во взрослого человека, но и в нейрофизиологическом. Конфликтные ситуации, эмоциональные стрессы, хронические заболевания, эндокринные расстройства, недостаток движения и прочие факторы являются во многом провокаторами развития вегетососудистой дистонии у подростков. В зрелом возрасте особую роль в запуске механизмов ВСД играют гормональные перестройки организма. Вот почему женская половина населения планеты страдает от ВСД гораздо чаще мужской. Предродовой период, беременность, период климакса, все это, являясь переломными моментами в жизни женщины, может стать отправной точкой для мобилизации проявления симптомов вегетососудистой дистонии. Особенно неблагоприятна вегетососудистая дистония при беременности, когда даже незначительные отклонения в здоровье женщины обязательно сказываются на состоянии плода. Это же справедливо и в отношении наличия лишнего веса, который может стать провокатором проявлений дистонии. Увеличение массы тела приводит к развитию гипертонии, что в свою очередь является дополнительной нагрузкой на сердечнососудистую систему. Развитие вегетососудистой дистонии в данном случае затрагивает людей совершенно разного возраста и пола.
Вот почему женская половина населения планеты страдает от ВСД гораздо чаще мужской. Предродовой период, беременность, период климакса, все это, являясь переломными моментами в жизни женщины, может стать отправной точкой для мобилизации проявления симптомов вегетососудистой дистонии. Особенно неблагоприятна вегетососудистая дистония при беременности, когда даже незначительные отклонения в здоровье женщины обязательно сказываются на состоянии плода. Это же справедливо и в отношении наличия лишнего веса, который может стать провокатором проявлений дистонии. Увеличение массы тела приводит к развитию гипертонии, что в свою очередь является дополнительной нагрузкой на сердечнососудистую систему. Развитие вегетососудистой дистонии в данном случае затрагивает людей совершенно разного возраста и пола.
Симптомы вегето-сосудистой дистонии.
Порой симптоматика вегетососудистой дистонии похожа на проявление сразу нескольких заболеваний одновременно. Это скачки артериального давления, головные боли, головокружения, сердцебиение и сердечные перебои, обмороки, ощущение волны жара, озноб, ледяные руки и ноги, повышенная потливость. Человек становится метеозависимым. Больные жалуются на слабость, быструю утомляемость, вялость, колебания температура тела (может опуститься до 35 градусов и подняться до 37 с лишним). Нередко расстройство пищеварения, боли в области живота, разнообразные боли во всем теле, особенно в момент глубокого вздоха. Имеется и совсем специфическая симптоматика: человек не может ездить в метро, долго стоять в очереди, сидеть в кресле у парикмахера, из-за того, что резко ощущает приступ беспокойства или не может справиться с нетерпением, беспричинной яростью или внезапным ощущением тяжести в руках и ногах. Однако самым неприятным моментом во всем этом является появление внезапной панической атаки с сильными болями в области сердце и под лопаткой, сопровождающиеся чувством смертельного ужаса.
Понятно, что имея такие симптомы, человек обращается за помощью к врачам. Начиная от терапевта и заканчивая психиатром. Но найти причину недугов бывает не так легко. Только после тщательного обследования врачи могут вынести вердикт: «Вегетососудистая дистония». А именно, не согласованная работа сосудов в момент сокращения и расслабления. Тем самым происходит нарушение в снабжении кровью и кислородом внутренних органов, что и вызывает сбой в их работе. Поэтому при вегетососудистой дистонии нет смысла лечить какой-то один орган, здесь необходим комплексный подход ко всему организму в целом. И в первую очередь лечение будет зависеть от самого больного.
Начиная от терапевта и заканчивая психиатром. Но найти причину недугов бывает не так легко. Только после тщательного обследования врачи могут вынести вердикт: «Вегетососудистая дистония». А именно, не согласованная работа сосудов в момент сокращения и расслабления. Тем самым происходит нарушение в снабжении кровью и кислородом внутренних органов, что и вызывает сбой в их работе. Поэтому при вегетососудистой дистонии нет смысла лечить какой-то один орган, здесь необходим комплексный подход ко всему организму в целом. И в первую очередь лечение будет зависеть от самого больного.
Течение ВСД
В большинстве случаев без провоцирующих факторов заболевание имеет латентный (бессимптомный) характер. Однако под воздействием неблагоприятных условий и перегрузок нередки проявления кризов. Кризовый всплеск активности болезни протекает тяжелее у людей старшего возраста, особенно у тех, кто страдает сопутствующими заболеваниями. Во многих случаях криз является результатом давно копившихся составляющих, а поэтому нередки случаи проявления большого количества симптомов одновременно.
Диагностика вегетососудистой дистонии значительно затруднена уже по определению. ВСД – это комплекс всевозможных проявлений различной природы. Поэтому строго классифицировать те или иные симптомы как присущие исключительно вегетососудистой дистонии просто невозможно. Нет в арсенале врачей и специальных приборов или особого оборудования, которое с большой долей вероятности определяло бы наличие расстройства. Безусловно, классические методы обследования важнейших органов и систем организма являются огромной помощью врачу-диагносту, но, основываясь лишь только на этих результатах, окончательно определить природу тех или иных симптомов довольно сложно. Часто комплексное сочетание тех или иных факторов вместе с результатами многосторонних анализов дают основание поставить диагноз.
Для правильной постановки диагноза врачу необходимо иметь на руках результаты различных исследований, важнейшие из которых: электрокардиография, эхография, реоэнцефалография, реовазография, магнитно-резонансная томография. Важны тщательные обследования и заключения следующих специалистов: лор, невролог, эндокринолог, окулист и даже психотерапевт. Опыт и история наблюдений за состоянием здоровья ближайших родственников тоже может помочь при определении причин вегето-сосудистой дистонии.
Важны тщательные обследования и заключения следующих специалистов: лор, невролог, эндокринолог, окулист и даже психотерапевт. Опыт и история наблюдений за состоянием здоровья ближайших родственников тоже может помочь при определении причин вегето-сосудистой дистонии.
Классическое лечение ВСД
Чаще всего в самом начале развития ВСД не требуется применять сильнодействующие медикаменты. Достаточно обойтись лишь природными седативными средствами, такими как боярышник, зверобой и валериана. Для психостимуляции применяют природные препараты на основе трав заманихи или лимонника. Из физиотерапевтических мероприятий в случае вегетососудистой дистонии назначают: ЛФК, классический массаж, иглоукалывание (рефлексотерапия) и водные процедуры (бассейн). Также врачи настоятельно рекомендуют больным ВСД покидать пределы города в качестве профилактических мер. Смена остановки очень полезно сказывается как на общем самочувствии, так и на нервно-эмоциональном здоровье тоже. Общение со специалистом-психологом также может оказать благотворное влияние на эмоциональное состояние больного.
При медикаментозном лечении вегетососудистой дистонии основное внимание сосредоточивается на устранении сопутствующих заболеваний, вирусной или хронической природы. Важно также отследить работу эндокринной системы организма, в частности щитовидной железы. При необходимости проводится комплексное лечение с привлечением гормональных препаратов. При кризовой форме проявления дистонии назначаются препараты, регулирующие деятельность сердца, успокоительные и витамины (особенно витамины группы B). Из более сильных препаратов назначают антидепрессанты, седативные средства, препараты для улучшения кровообращения и др.
Профилактика ВСД
Правильное питание и здоровый образ жизни – избитые фразы, но, в случае с вегетососудистой дистонией, приобретающие особое значение. Даже люди, имеющие наследственную предрасположенность к заболеванию, имеют все шансы одержать победу над недугом, если противопоставят дистонии крепкое здоровье и хороший полноценный сон. Большинство врачей сходятся во мнение, что регулярные пешие прогулки (а лучше пробежки) укрепляют не только сердечную мышцу, но и иммунитет. Частое «общение с природой» благотворно действует на восстановительные функции организма, ускоряя их и восстанавливая.
Большинство врачей сходятся во мнение, что регулярные пешие прогулки (а лучше пробежки) укрепляют не только сердечную мышцу, но и иммунитет. Частое «общение с природой» благотворно действует на восстановительные функции организма, ускоряя их и восстанавливая.
ГБУЗ «Центр медицинской профилактики» министерства здравоохранения Краснодарского края
Вегето-сосудистая дистония: причины, симптомы, лечение
Диагностика ВСД
Подозрения на диагноз вегето-сосудистой дистонии являются весомым поводом провести полное обследование организма. В данном случае для точного определения причин проявления симптомов ВСД следует провести комплексные исследования организма.
Дело в том, что ВСД имеет более 150 различных симптомов (мы обозначили лишь те, которые проявляются наиболее часто). После выявления первопричины специалист назначит дополнительные исследования и разработает индивидуальный курс лечения.
Лечение ВСД
Курс лечения ВСД – это широкий комплекс мероприятий. К сожалению, вегето-сосудистая дистония является сложным симптоматическим «набором», который требует профессионального и всестороннего подхода.
Наряду с лечением основного заболевания, пациенту также потребуется изменить привычный образ жизни (отказаться от вредных привычек, создать благоприятную социально-психологическую ситуацию, восстановить режим бодрствования и сна). Это позволит сократить количество ежедневных стрессовых ситуаций.
Лечение ВСД подразумевает три основных направления:
- Лечение основного заболевания (преимущественно соматического характера), которое является причиной проявления дистонических симптомов.
- Посещение психотерапевта, в процессе которого пациенту помимо применения индивидуальных техник также может быть назначено медикаментозное лечение (как правило, назначаются антидепрессанты или легкие седативные препараты, действие которых направлено на снижение уровня тревожности).
- Восстанавливающие техники, среди которых часто применяются: массаж, дыхательная гимнастика, витаминотерапия, иглоукалывание, занятия йогой.

Устранить симптомы ВСД и вылечить заболевание, скрывающееся за ними, можно. Это позволит значительно повысить качество жизни. Согласитесь, что ощущать себя здоровым намного лучше, чем пребывать в состоянии слабости и испытывать боль, которая проявляется во всех частях тела. Отличное состояние всех органов и о в целом организма стоит того, чтобы найти время и пройти обследование у всех профильных специалистов.
Обновленная информация о детских дистониях: этиология, эпидемиология и лечение
Резюме
Дистония — это двигательное расстройство, характеризующееся длительными мышечными сокращениями, вызывающими скручивающие, повторяющиеся и шаблонные движения или неправильные позы. Дистония — одно из наиболее часто наблюдаемых двигательных расстройств в клинической практике как у взрослых, так и у детей. Он классифицируется на основе этиологии, возраста появления симптомов и распределения пораженных участков тела.
Этиология
Этиология детской дистонии весьма неоднородна.Существует множество различных генетических синдромов и несколько причин симптоматических синдромов. Дистония может быть вторичной по отношению практически к любому патологическому процессу, поражающему двигательную систему, и особенно базальные ганглии.
Классификация
Этиологическая классификация различает первичную дистонию без идентифицированной экзогенной причины или признаков нейродегенерации и вторичных синдромов.
Лечение
Лечение большинства форм дистонии симптоматическое и включает в себя лекарственные препараты (системные или очаговые методы лечения, такие как ботулинический токсин) и хирургические процедуры.Есть несколько лекарств, в том числе антихолинергические, дофамин-блокирующие и истощающие агенты, баклофен и бензодиазепины. У пациентов с нарушениями синтеза дофамина лечение L-допа может быть очень полезным. Лечение ботулиническим токсином может быть полезным в борьбе с наиболее ограничивающими симптомами сегментарной или фокальной дистонии. Длительная электрическая стимуляция внутреннего бледного шара оказывается особенно успешной у детей, страдающих генерализованной дистонией.
У пациентов с нарушениями синтеза дофамина лечение L-допа может быть очень полезным. Лечение ботулиническим токсином может быть полезным в борьбе с наиболее ограничивающими симптомами сегментарной или фокальной дистонии. Длительная электрическая стимуляция внутреннего бледного шара оказывается особенно успешной у детей, страдающих генерализованной дистонией.
Ключевые слова: двигательные расстройства, детская дистония, первичные дистонии, вторичные дистонии
Введение
Дистония — это двигательное расстройство, характеризующееся устойчивыми мышечными сокращениями, вызывающими скручивание, повторяющиеся и шаблонные движения или аномальные позы.Дистония имеет широкий клинический спектр: от минимальных или доброкачественных самоограничивающихся признаков до тяжелых случаев. 1 — 3 Дистония классифицируется на основе: (i) этиологии, (ii) возраста появления симптомов и (iii) распределения пораженных участков тела. Этиология детской дистонии неоднородна (). 4 Этиологическая классификация различает первичную дистонию без идентифицированной экзогенной причины или признаков нейродегенерации и вторичных синдромов.Первичные заболевания можно разделить на чистые, плюс и пароксизмальные. В первичных чистых формах дистония является единственным признаком заболевания (за исключением тремора), а причина либо генетическая, либо неизвестная. Первичные плюсовые синдромы характеризуются дистонией, связанной с дополнительным двигательным расстройством (например, миоклонусом или паркинсонизмом). Наконец, первичные пароксизмальные синдромы включают состояния, характеризующиеся аномальными движениями, включая дистонию, возникающую в виде коротких эпизодов с нормальным состоянием между ними.Вторичные дистонические формы включают синдромы, при которых дистония является ярким признаком наследственно-дегенеративного состояния или когда дистония вызвана экзогенными факторами (например, перинатальная травма, лекарства, опухоль головного мозга, инфекции).
Таблица 1
Классификация дистоний по этиологии
| Первичная | |
|---|---|
| • Первичная чистая | Дистония является единственным клиническим признаком (кроме тремора), и нет никакой идентифицируемой экзогенной причины или другое наследственное или дегенеративное заболевание |
| • Первичная плюс | Дистония является заметным признаком, но связана с другим двигательным расстройством, например миоклонусом или паркинсонизмом.Нет свидетельств нейродегенерации. |
| • Первичная пароксизмальная | Дистония возникает короткими эпизодами с нормальным состоянием между ними. Эти расстройства классифицируются как идиопатические (часто семейные, хотя встречаются и спорадические случаи) и симптоматические из-за множества причин |
| Вторичные | Дистония является симптомом определенного неврологического состояния, такого как очаговое поражение мозга, воздействие лекарств. или химические вещества |
| Наследственная дегенеративная | Дистония — это особенность, среди других неврологических признаков, наследственно-дегенеративного или метаболического расстройства |
Вторая ось классификации различает начало у детей (<18 лет) и начало у взрослых (> 19 лет) кейсы (). Наконец, третья ось, основанная на соматическом распределении дистонии, различает фокальные, сегментарные, многоочаговые, генерализованные и односторонние (гемидистония) формы ().
Наконец, третья ось, основанная на соматическом распределении дистонии, различает фокальные, сегментарные, многоочаговые, генерализованные и односторонние (гемидистония) формы ().
Таблица 2
Классификация дистоний по возрасту начала
| Раннее начало (варьируется от 20 до 30 лет) | Обычно начинается с конечности и часто распространяется на другие конечности и туловище |
| Позднее начало | Обычно начинается с шеи (включая гортань), черепных мышц или одной руки.Имеет тенденцию оставаться локализованной с ограниченным прогрессированием в соседние мышцы |
Таблица 3
Классификация дистоний по распределению
| Очаговая | Отдельная область тела |
| Сегментарная | Смежные области тела |
| Многоочаговая | Несмежные области тела |
| Обобщенный | Обе ноги и хотя бы одна другая часть тела |
| Гемидистония | Половина тела |
Дистонические синдромы относятся к наиболее часто наблюдаемым двигательным расстройствам в клинической практике как у взрослых, так и у детей, с распространенностью от 2 до 50 случаев на миллион для дистонии с ранним началом (<26 лет) и от 30 до 7320 случаев на миллион для дистонии с поздним началом (> 26 лет). 5 — 7
5 — 7
В этом обзоре мы сосредоточимся на дистонии, которая поражает детей, произвольно ограничиваясь лицами моложе 18 лет. Мы обсудим первичные дистонии с педиатрическим началом, разделенные на «чистые» дистонии (DYT1, DYT6, DYT13, DYT17), «дистонии плюс», связанные с миоклонусом (DYT11 и DYT15), и «дистонии плюс», связанные с паркинсонизмом (DYT5). , DYT12, DYT16). Наконец, мы включим наиболее частые вторичные дистонии.
Синдромы первичной чистой дистонии
В чистом виде дистония является единственным клиническим признаком (кроме тремора).Детская первичная чистая дистония — редкое заболевание; сюда входят семейные и спорадические случаи. Было картировано семь локусов для первичной дистонии, начинающейся у детей, включая DYT1, DYT4, DYT6, DYT13 и DYT17 (). Среди них на данный момент известны только два причинных гена, DYT1 / TOR1A и DYT6 / THAP1. 8
Таблица 4
Молекулярная классификация непароксизмальных дистоний, начинающихся у детей
| Ген | Локус | Обозначение | Продукт гена | Тип |
|---|---|---|---|---|
| С аутосомно-доминантным наследованием | ||||
| DYT1 | 9q34 | Идиопатическая торсионная дистония (ITD) | Торсин A ( TOR1A ) | Первичный чистый |
| DYT4 | Неизвестно | Идиопатическая торсионная дистония | Неизвестная торсионная дистония Неизвестная торсионная дистония 4949||
| DYT5 | 14q22. 1 1 | Дефицит GTPCH | GTP-циклогидролаза 1 ( GCh2 ) | Первичный плюс |
| DYT6 | 8p11–21 | Подростковое / раннее взрослое начало | N-концевой домен THAP ( THAP1 ) | Первичный чистый |
| DYT11 | 7q21 AD | Миоклоническая дистония | ε-саркогликан ( SGCE ) | Первичный плюс |
| DYT12 | 9q13 AD | Rapid-onset + / K + -ATPase ( ATP1A3 ) | Основной плюс | |
| DYT13 | 1p36.13–36,32 | Дистония с ранним началом с поражением шейного отдела черепа и верхних конечностей | Неизвестно | Первичная чистая |
| DYT15 | 18p11 | Миоклоническая дистония, чувствительная к алкоголю | Неизвестно | |
| DYT16 | 2q31.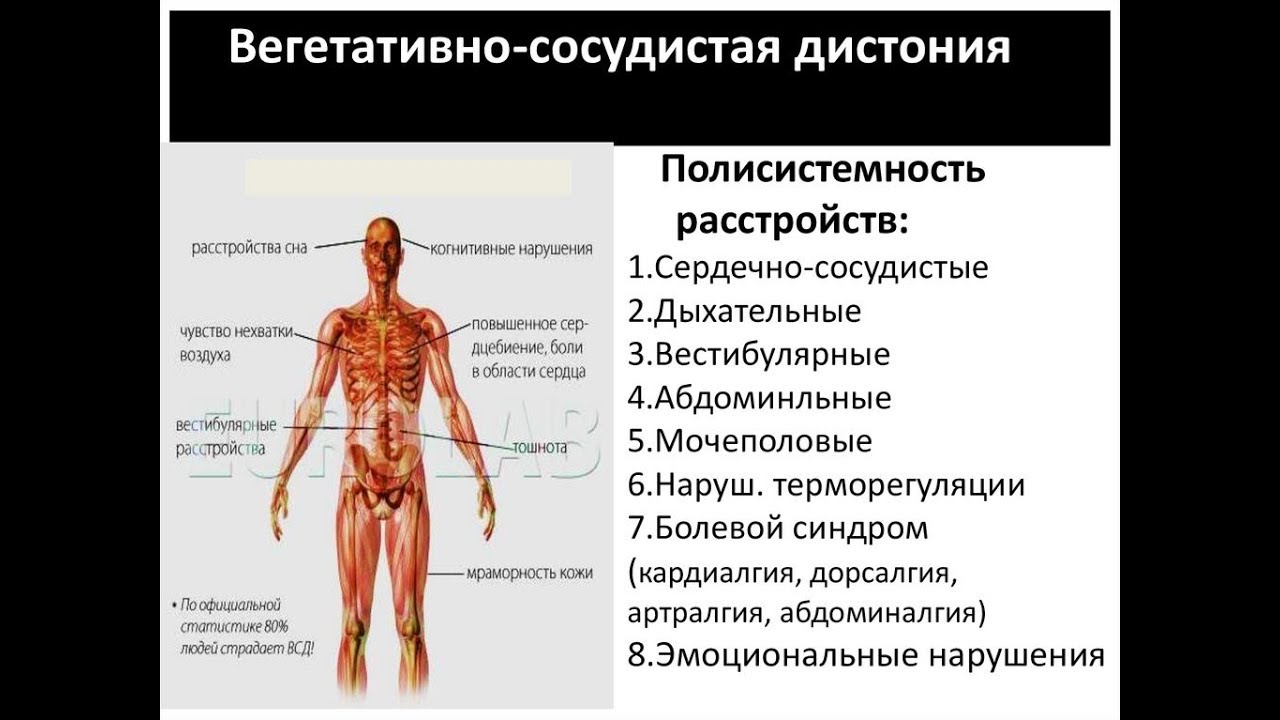 2 2 | Дистония-паркинсонизм с молодым началом | Киназа PKR ( PRKRA ) | Первичная плюс |
| DYT17 | 20p11.22-q13.12 | Аутосомно-рецессивный первичный очаговый TD | Неизвестно | Первичный чистый |
| С Х-сцепленным наследованием | ||||
| DYT3 | Xq13.1 | Х-сцепленная дистония / паркинсонизм | Фактор 1, связанный с TATA-связывающим белком ( TAF1 ) | Первичный плюс |
| Xq13 | X-связанный синдром дистонии-глухоты (синдром Мора-Транебьерга) | Импортная транслоказа внутренней мембраны митокондрий Tim8A ( TIMM8A ) | ||
DYT1 (
TOR1A ): Дистония с ранним началом Дистония с ранним началом, вызванная мутацией DYT1 , представляет собой наиболее частую и наиболее тяжелую форму первичной дистонии.На его долю приходится 16–53% случаев детской дистонии (POD) среди нееврейского населения и 80–90% пациентов среди еврейского населения ашкенази. 9 — 11
9 — 11
Рассчитанная частота заболевания среди еврейского населения ашкенази составила 1: 3000–1: 9000. Среди нееврейского населения частота примерно в пять раз ниже. 12 , 13
Обычно дистония DYT1 начинается в детстве или подростковом возрасте. Первоначальным симптомом обычно является дистония очагового действия, затрагивающая одну конечность (писчая дистония, ходячая дистония с инверсией или выворотом стопы).Дистония впоследствии распространяется на другие области тела, становится менее специфичной и может также присутствовать в состоянии покоя. Пациенты с дистонией ног в начале болезни обычно имеют более раннее начало (средний возраст 9 лет) и генерализованную дистонию через несколько месяцев или лет. Пациенты с дистонией руки имеют более позднее начало (средний возраст 15 лет), и генерализация дистонии встречается реже. Дистония также может начаться в шее или черепных мышцах. 14
В целом, до 65% пациентов с дистонией DYT1 имеют генерализованную дистонию, при которой чаще всего поражаются конечности.Черепные мышцы поражаются реже, примерно у 11–18% пациентов. Пораженные члены семьи могут иметь начало у взрослых и чаще с поражением шеи. Сообщалось о необычных фенотипах, таких как изолированный блефароспазм и односторонняя миоклоническая дистония. 15
У некоторых пациентов с дистонией DYT 1 может наблюдаться резкое ухудшение дистонии, называемое «status dystonicus», в течение болезни. 16 Продукт гена DYT1 , TorsinA, является членом суперсемейства АТФаз AAA1, связанных с различными клеточными активностями.Он выполняет важные функции, связанные с деградацией белков, переносом мембран, слиянием везикул и перемещением органелл, динамикой цитоскелета и правильным сворачиванием белков. 17
TorsinA экспрессируется почти повсеместно, и его экспрессия в головном мозге ограничена нейронами, где он связан с эндоплазматическим ретикулумом (ER). В клеточных моделях, экспрессирующих патогенную делецию GAG, мутантный TorsinA перераспределяется из просвета ER в ядерную оболочку (NE). 18 Эти клетки также обнаруживают аномальную морфологию и утолщение NE, включая образование включений мутовидной мембраны, которые, по-видимому, происходят из ER и NE. Эти включения связаны с везикулярным транспортером моноаминов VMAT2, открытие, которое может функционально связать TorsinA с дофаминергической системой. 19 Кроме того, было обнаружено, что TorsinA регулирует клеточный перенос переносчика дофамина и других мембраносвязанных белков. Также было показано, что мутант TorsinA вмешивается в процессы цитоскелета, которые могут влиять на развитие нейрональных путей в головном мозге, и что он преждевременно разрушается как протеасомными путями, так и путями макроаутофагии. 20 Недавно было показано, что TorsinA контролирует стабильность белков синаптических везикул и влияет на оборот синаптических везикул в нейронах. 21
В клеточных моделях, экспрессирующих патогенную делецию GAG, мутантный TorsinA перераспределяется из просвета ER в ядерную оболочку (NE). 18 Эти клетки также обнаруживают аномальную морфологию и утолщение NE, включая образование включений мутовидной мембраны, которые, по-видимому, происходят из ER и NE. Эти включения связаны с везикулярным транспортером моноаминов VMAT2, открытие, которое может функционально связать TorsinA с дофаминергической системой. 19 Кроме того, было обнаружено, что TorsinA регулирует клеточный перенос переносчика дофамина и других мембраносвязанных белков. Также было показано, что мутант TorsinA вмешивается в процессы цитоскелета, которые могут влиять на развитие нейрональных путей в головном мозге, и что он преждевременно разрушается как протеасомными путями, так и путями макроаутофагии. 20 Недавно было показано, что TorsinA контролирует стабильность белков синаптических везикул и влияет на оборот синаптических везикул в нейронах. 21
Первичные дистонии с ранним началом, не связанные с DYT1
Большая группа пациентов с ПОЛ не связана с мутацией DYT1 . DYT6 дистония была описана в двух семьях германо-меннонитского происхождения. 22 Фенотип определяется как смешанный, потому что он отличается от типичного педиатрического начала: варьирующий возраст в начале от младенчества до взрослого возраста (диапазон 5–38 лет; среднее значение 18.6 лет), выраженное поражение черепных и шейных мышц, генерализация дистонии наблюдается примерно у половины пациентов. 23 Продукт гена DYT6 , Танатос-ассоциированный белок 1 (THAP1), является членом семейства клеточных факторов, разделяющих высококонсервативный ДНК-связывающий домен THAP1, который представляет собой атипичный цинковый палец и может регулировать эндотелиальные клетки. распространение. 24 Предполагаемый механизм заболевания заключается в том, что мутации DYT6 могут нарушать связывание THAP1 с ДНК и вызывать нарушение регуляции транскрипции. Недавно было продемонстрировано, что между THAP1 и промотором TOR1A происходит физическое взаимодействие, которое устраняется патогенными мутациями. 25
Недавно было продемонстрировано, что между THAP1 и промотором TOR1A происходит физическое взаимодействие, которое устраняется патогенными мутациями. 25
Эта связь, вероятно, не просто простое взаимодействие промоторов THAP1-Tor1A, но, вероятно, также включает другие гены. Интересно, что THAP1 также связывается с промоторной областью транскрипта TAF1, который участвует в форме первичной плюс дистонии (Х-сцепленная паркинсонизм с началом Х-хромосомы). 26
Другой редкой причиной раннего начала первичной дистонии является мутация DYT13 , которая была обнаружена в большой итальянской семье. 27 Преобладающим фенотипом является детская дистония верхней части тела с частым поражением черепно-шейной области и относительно доброкачественным течением.
Первичная дистония DYT17 описана как пример аутосомно-рецессивного POD. 28 , 29 Локус для DYT17 был назначен на хромосому 20.
Первичные плюсовые синдромы
Дистония является заметным признаком дистонии плюс синдромов, но она связана с другими двигательными расстройствами (например, myoclonus или паркинсонизм) без признаков нейродегенерации.Выделяют три клинически определяемых состояния: допа-зависимая дистония ( DRD; DYT5 ), миоклоническая дистония (MD; DYT11 ) и быстро развивающаяся дистония-паркинсонизм (RDP; DYT12 ), которые характеризуются выраженным фенотипическая гетерогенность.
DRD или болезнь Сегавы
DRD или болезнь Сегавы в основном возникает у детей из-за дефектов синтеза дофамина. Обычный возраст начала заболевания колеблется от 4 до 6 лет. Соотношение самок и самцов — 2.5: 1. 30 Сообщалось о распространенности от 0,5 до 1 на миллион, но, вероятно, она недооценена, поскольку пенетрантность низкая, а атипичные случаи часты. 31 Начало часто коварное, с утомляемостью, неуклюжей походкой и дистоническими позами, часто ограниченными одной стопой. 32 , 33
32 , 33
У детей старше 10 лет первыми проявлениями могут быть дистония верхних конечностей или постуральный тремор. 34 Обычно присутствуют легкие признаки паркинсонизма, такие как ригидность конечностей и туловища и гипомимия.Очень характерно прогрессирующее усиление дистонии в течение дня и заметное уменьшение, а иногда и полное исчезновение после сна. Расстройство прогрессирует и может привести к тяжелой инвалидности. Поражение других частей тела может произойти быстро или занять до десяти лет. Когнитивные функции сохранены.
Дистония и / или паркинсонизм обычно показывают быстрый, выраженный и устойчивый ответ на низкие дозы (3-5 мг / кг / день) L-допа в сочетании с ингибитором периферического декарбоксилирования, помогающим установить клинический диагноз.Ответ не зависит от задержки в начале лечения. Дефицит аутосомно-доминантной гуанозинтрифосфатциклогидролазы (GTPCH) ( DYT5 ) является наиболее частым ферментативным дефектом, приводящим к болезни Сегавы. Почти 75% случаев индекса DRD большой европейской серии показали этот дефект. 35
Сообщалось об аутосомно-рецессивных формах GTPCH. 36 Аутосомно-рецессивные формы DRD (ArDRD) также возникают в результате гомозиготных или сложных гетерозиготных мутаций тирозингидроксилазы (TH) и сепиаптеринредуктазы (SPR). 37 При ArDRD фенотип обычно более тяжелый, включая когнитивные нарушения и задержку развития, и его также можно назвать инфантильным энцефалопатическим фенотипом (IEP). 3 Ответ на лечение L-допа при IEP отличается от болезни Сегавы. Только в редких случаях низкие дозы L-допы могут привести к быстрому улучшению симптомов. У большинства этих пациентов низкие дозы L-допы могут вызывать выраженные хореические и / или дистонические движения. У большинства этих пациентов необходимо очень медленное титрование дозы, и часто невозможно достичь эффективных терапевтических уровней L-допа. Даже низкие дозы могут вызывать заметные хореические и / или дистонические движения. Прогноз ИЭП отличается от прогноза болезни Сегавы даже в тех случаях, когда частично отвечает дофаминергическая терапия; Часто сообщается о неуклюжести и легкой умственной отсталости.
Даже низкие дозы могут вызывать заметные хореические и / или дистонические движения. Прогноз ИЭП отличается от прогноза болезни Сегавы даже в тех случаях, когда частично отвечает дофаминергическая терапия; Часто сообщается о неуклюжести и легкой умственной отсталости.
Недавно у некоторых пациентов с ИЭП, вызванным мутациями в гене, кодирующем переносчик допамина ( SLC6A3 ), было зарегистрировано новое состояние, называемое синдромом дефицита переносчика дофамина (DTDS). Исследования нейротрансмиттеров спинномозговой жидкости показали повышенное соотношение HVA / HIAA.Сканирование переносчика дофамина с помощью однофотонной эмиссионной компьютерной томографии (DaTSCAN SPECT) показывает полную потерю активности переносчика дофамина в базальных ядрах. 38
Миоклоническая дистония (M-D) (
DYT11 )Заболевание начинается в детском или подростковом возрасте. Симптомом обычно является миоклонус, поражающий верхнюю часть тела (шею, туловище, конечность) с преобладанием проксимальных мышц. Также могут быть затронуты черепные мышцы, в том числе мышцы гортани. 39 — 43 Дистония связана с миоклонусом более чем у половины пациентов и обычно вызывает незначительную инвалидность или не вызывает ее вовсе. Дистония обычно поражает шею или руки (кривошея, судорога писателя). 40 , 41 , 44 — 46 Заметное улучшение двигательных симптомов (в основном миоклонуса) после приема алкоголя и наличия психических симптомов, таких как обсессивно-компульсивные расстройства, злоупотребление алкоголем часто сообщается о депрессии и тревоге. 44 , 47 — 49 Течение болезни, как правило, доброкачественное, но исходы у пациентов, даже в пределах одной семьи, сильно различаются. 41
M-D вызывается гетерозиготными мутациями гена эпсилон-саркогликана ( SGCE ). Выявлено более 50 различных гетерозиготных мутаций SGCE . 50 Считается, что эти мутации вызывают потерю функции и не позволяют провести корреляцию генотип-фенотип.Однако заметные различия в степени тяжести заболевания предполагают влияние факторов окружающей среды или генетических модификаторов. Геномной особенностью SGCE является материнский геномный импринтинг. 51
Выявлено более 50 различных гетерозиготных мутаций SGCE . 50 Считается, что эти мутации вызывают потерю функции и не позволяют провести корреляцию генотип-фенотип.Однако заметные различия в степени тяжести заболевания предполагают влияние факторов окружающей среды или генетических модификаторов. Геномной особенностью SGCE является материнский геномный импринтинг. 51
Дистония-паркинсонизм с быстрым началом (RDP,
DYT12 ) Заболевание характеризуется внезапной дистонией и паркинсонизмом. 52 , 53 В своей генетической форме тип наследования аутосомно-доминантный с пониженной пенетрантностью, и сообщалось о мутациях de novo. 54 , 55 Одной из моногенетических причин RDP является мутация в гене альфа-субъединицы Na + / K + -АТФазы 3 (a3; ATP1A3 ). Хотя зарегистрировано всего несколько семей и единичные случаи с этим заболеванием, клинические особенности делают это заболевание особенно интересным. Расстройство обычно начинается в возрасте от 15 до 45 лет. Отличительными признаками являются резкое появление двигательных симптомов в течение от нескольких минут до 30 дней с вовлечением рострокаудальной области и выраженными бульбарными находками.Начало часто следует за физическим или эмоциональным стрессом, приводящим к стойкой неврологической инвалидности. 54 Большинство пациентов остаются стабильными или демонстрируют небольшое улучшение через годы после резкого появления симптомов. Заболевание передается по аутосомно-доминантному признаку с переменной выраженностью и пониженной пенетрантностью. Мутации в гене ATP1A3 (хромосома 19q13), связанном с NA + / K + ATPase alpha3, являются причиной нарушения. 54 , 55 RDP — первое известное дистоническое состояние, связанное с аномалией мембранного ионного канала.Все исследования остаются отрицательными, за исключением низкого уровня HVA в спинномозговой жидкости, что свидетельствует о пресинаптическом дефекте нигростриатных путей. 56
56
Недавно был идентифицирован новый ген PRKRA (протеинкиназа, интерферон-индуцибельный двухцепочечный РНК-зависимый активатор) для молодой дистонии-паркинсонизма ( DYT16 ). 57
Другие первичные генерализованные дистонии
Х-связанная дистония глухоты
Сообщалось о нескольких семьях с синдромом Х-связанной дистонии-глухоты (также называемым синдромом Мора – Транебьерга). 58 , 59 Заболевание характеризуется началом сенсоневральной глухоты обычно в возрасте до 2 лет. Тяжелая прогрессирующая генерализованная дистония начинается примерно в 7-летнем возрасте. Сообщалось о случаях развития дистонии уже в возрасте 30 лет, что позволяет предположить, что клинические признаки имеют более широкий спектр, чем считалось ранее. 60 , 61 Большинство испытуемых приковывались к инвалидной коляске в возрасте от 9 до 22 лет.Умственная отсталость, нарушение зрения из-за корковой слепоты или связанное с нейропатией зрительного нерва, 62 вовлечение кортикоспинального тракта и психиатрические проявления (раздражительность, беспокойство, паранойя) могут быть ассоциированными признаками. Ген-кандидат для этого расстройства, названный DDP (пептид глухоты / дистонии) или TIMM8A (вероятно, кодирующий импортируемую в митохондрии субъединицу транслоказы внутренней мембраны Tim8 A, цинк-связывающий белок), был идентифицирован на хромосоме Xq22. 63 Белок (белок DDP) расположен в межмембранном пространстве митохондрий, поэтому мутированный белок DDP может нарушить функцию митохондрий. 64 Сообщалось о нескольких мутациях, включая интронные. 65 По крайней мере, в одной из описанных мутаций самки-носители также страдают дистонией, но, в отличие от мужчин, она описывается как очаговая дистония. 66
ГАМКергические вещества, такие как клоназепам и гаммагидроксимасляная кислота, считаются полезными. 60
60
Преходящая идиопатическая дистония младенчества
Преходящая дистония младенчества обычно проявляется в возрасте до 5 месяцев. 67 , 68 У пораженных младенцев аномальные позы обычно ограничиваются одной верхней или нижней конечностью. Иногда в позы могут вовлекаться туловище, руки или одна сторона тела. 69 В положении лежа младенец часто поддерживает форсированную пронацию предплечья, используя тыльную сторону кисти в качестве опоры. В нижней конечности ступни могут находиться в эквиноварусии. Позы могут быть прерывистыми. 70 В остальном неврологическое обследование и развитие в норме.Двигательное расстройство самопроизвольно исчезает к первому дню рождения. Семейные случаи известны. 67 , 68 Легкие формы могут быть частыми, и только в наиболее выраженных случаях может потребоваться медицинская помощь. Ключом к диагнозу является наблюдение, что дистоническая поза исчезает, когда младенец выполняет целенаправленные движения пораженной конечностью. Этиология пока неизвестна.
При типичных клинических признаках дополнительные исследования (нейровизуализация, лабораторные исследования) не требуются.
Первичные пароксизмальные синдромы
Пароксизмальные дискинезии относятся к приступам аномальных движений и поз (дистонических, хореоатетоидных или их комбинации) с возвращением к нормальному состоянию между эпизодами. 71
Можно выделить три основные категории: пароксизмальная кинезигенная дискинезия (PKD / DYT10), пароксизмальная некинезигенная дискинезия (PNKD1; DYT8) и пароксизмальная дискинезия, вызванная физической нагрузкой (PED; DYT18). 72 В ДОК продолжительность атак варьируется от секунд до часов, а частота эпизодов варьируется от одного в год до сотен в день.PKD характеризуется дистонией или хореодистонией, вызванной внезапной сменой положения, обычно переходящего из положения сидя в положение стоя. PNKD характеризуется спонтанными приступами в покое, которые имеют тенденцию к более дистоническому характеру, хотя могут возникать хорея, атетоз и баллизм. Эпизоды могут длиться от секунд до нескольких часов и иметь длинные интервалы без приступов. Начало в детстве с тенденцией к уменьшению приступов с возрастом. 73 Симптомы могут быть спровоцированы алкоголем или кофеином и, в меньшей степени, никотином, возбуждением, усталостью, голодом и эмоциональным стрессом.Пароксизмальный хореоатетоз с клиническими характеристиками, подобными таковым для PNKD с ассоциированной спастичностью, был описан под термином аутосомно-доминантный пароксизмальный хореоатетоз / синдром спастичности (CSE), и была продемонстрирована связь с хромосомой 1p. 74 В PED приступы возникают после 10 или 15 минут непрерывных упражнений. Приступы обычно дистонические и чаще всего появляются в нижних конечностях после продолжительной ходьбы или бега. Приступы дистонии обычно прекращаются через 10–15 минут после прекращения упражнений. 75
PNKD характеризуется спонтанными приступами в покое, которые имеют тенденцию к более дистоническому характеру, хотя могут возникать хорея, атетоз и баллизм. Эпизоды могут длиться от секунд до нескольких часов и иметь длинные интервалы без приступов. Начало в детстве с тенденцией к уменьшению приступов с возрастом. 73 Симптомы могут быть спровоцированы алкоголем или кофеином и, в меньшей степени, никотином, возбуждением, усталостью, голодом и эмоциональным стрессом.Пароксизмальный хореоатетоз с клиническими характеристиками, подобными таковым для PNKD с ассоциированной спастичностью, был описан под термином аутосомно-доминантный пароксизмальный хореоатетоз / синдром спастичности (CSE), и была продемонстрирована связь с хромосомой 1p. 74 В PED приступы возникают после 10 или 15 минут непрерывных упражнений. Приступы обычно дистонические и чаще всего появляются в нижних конечностях после продолжительной ходьбы или бега. Приступы дистонии обычно прекращаются через 10–15 минут после прекращения упражнений. 75
Пациенты с мутациями гена SLC2A1 (ген, кодирующий транспортер Glut 1) могут иметь PED, который может быть связан с эпилепсией и / или умственной отсталостью. 76 — 78
Список локусов сцепленных генов, вызывающих фенотипы пароксизмальной дискинезии, быстро растет, хотя гены большинства этих состояний все еще остаются неидентифицированными. Пароксизмальные дискинезии наследуются по аутосомно-доминантному признаку. 79 PNKD вызывается мутациями в гене PNKD1 , кодирующем регулятор 1 миофибриллогенеза.При PKD локус заболевания был приписан перицентромерной области хромосомы 16 (16p11.2-q12.1). 80 Первый ген, вызывающий PKD, был недавно идентифицирован: трансмембранный белок пролинерих 2. 81 , 82 Ген PED расположен в коротком плече хромосомы 1 (1p31. 3 – p35). Ген SLC2A1 кодирует транспортер глюкозы 1 (Glut1). 77 Поскольку Glut1 доставляет глюкозу через гематоэнцефалический барьер, дискинезии могут быть результатом дефицита энергии в базальных ганглиях при физической нагрузке.
77 Поскольку Glut1 доставляет глюкозу через гематоэнцефалический барьер, дискинезии могут быть результатом дефицита энергии в базальных ганглиях при физической нагрузке.
Вторичные дистонии
Дистония часто ассоциируется с другими неврологическими симптомами в длинном списке заболеваний, и затем ее называют вторичной дистонией.
Дистония, вызванная структурным дефектом
Дистонический церебральный паралич и дистония с отсроченным началом не будут обсуждаться в этом обзоре. Гемидистония часто возникает из-за структурного поражения. Наиболее частой причиной гемидистонии является острая окклюзия сосудов любой этиологии, особенно поражающая полосатое тело и / или хвостатое тело. 83 Гемидистония чаще встречается у детей, чем у взрослых. 84 Гемидистония преобладает в верхних конечностях и очень часто сопровождается признаками пирамидного тракта. Первым неврологическим проявлением обычно является острый гемипарез. Дистония может появиться во время выздоровления гемипареза, но часто она становится очевидной только через несколько месяцев или даже лет. 84
Часто оказывается плохой ответ на лечение. Наилучшие результаты дают бензодиазепины и антихолинергические препараты.Может быть полезен ботулинический токсин. Хирургия, в основном глубокая стимуляция головного мозга (DBS) в определенных областях внутреннего бледного шара, кажется сейчас лечением выбора. 85
Наследственные дегенеративные синдромы
Наследственные дегенеративные синдромы составляют большую и разнородную группу заболеваний, при которых дистония может возникать с разной степенью тяжести и связана с другими аномалиями, включая умственную отсталость, судороги, атаксию, невропатию, атрофию зрительного нерва и взор. парез.Многие из этих расстройств являются аутосомно-рецессивными из-за наследственных дегенеративных или метаболических заболеваний и возникают в основном в детстве.
Глутаровая ацидурия и другие органические ацидемии с дистонией
Глутаровая ацидурия I типа (GAI), также называемая дефицитом глутарил-КоА-дегидрогеназы, представляет собой аутосомно-рецессивное заболевание, вызываемое мутациями в гене глутарил-КоА-дегидрогеназы ( GCDH), расположенном на хромосоме GCDH. 19п13.2. 86 , 87 GCDH — митохондриальный фермент, который играет ключевую роль в катаболизме лизина, гидроксилизина и триптофана.Сильное снижение или полное отсутствие активности GCDH вызывает накопление 3-гидроксиглутаровой кислоты и глутаровой кислоты в плазме, моче и спинномозговой жидкости, что может вызвать гибель нейронов из-за эксайтотоксичности, а также дисфункции митохондрий. 88
19п13.2. 86 , 87 GCDH — митохондриальный фермент, который играет ключевую роль в катаболизме лизина, гидроксилизина и триптофана.Сильное снижение или полное отсутствие активности GCDH вызывает накопление 3-гидроксиглутаровой кислоты и глутаровой кислоты в плазме, моче и спинномозговой жидкости, что может вызвать гибель нейронов из-за эксайтотоксичности, а также дисфункции митохондрий. 88
Частота ГАИ оценивается в 1 на 100 000 новорожденных. 89 Клинические проявления обычно появляются в возрасте от 5 до 14 месяцев, но легкие симптомы, такие как небольшая задержка моторики и гипотония, могут наблюдаться в более раннем возрасте.Макроцефалия при рождении или развившаяся позже в младенчестве присутствует примерно в 70% случаев. 90 , 91 В большинстве случаев заболевание начинается внезапно, с очаговыми припадками или генерализованными судорогами, рвотой и заторможенностью или летаргией, обычно после острого инфекционного заболевания. Позже появляются психомоторный регресс и дистонические или хореоатетотические движения. У некоторых пациентов начало незаметно с медленно развивающейся задержкой психомоторного развития, гипотонией и дистоническими позами до 16 лет. 92 — 95
Характерным неврологическим последствием является генерализованная дистония. Также может быть очевидна спастичность. Часто встречаются сложные речевые изменения с сочетанием признаков гиперкинетической дизартрии, анартрии и апраксии. Понимание языка намного лучше, чем выражение, что говорит о том, что познание относительно сохранено.
Течение расстройства непостоянно. ГАИ — излечимое состояние. В идеале лечение следует начинать до появления клинических симптомов.В таких случаях диета в сочетании с пероральным приемом L-карнитина и агрессивным лечением во время острых эпизодов интеркуррентного заболевания может предотвратить развитие клинических признаков или уменьшить их тяжесть. Однако в большинстве случаев болезнь продолжает прогрессировать поэтапно с чередой острых энцефалопатических кризов, вызванных инфекционными заболеваниями, вакцинацией и хирургическим вмешательством в младенчестве или детстве. Часто с возрастом дистония имеет тенденцию эволюционировать от мобильной к стационарной и ассоциироваться с паркинсонизмом. 96 В других случаях он остается неподвижным с тяжелыми двигательными и языковыми последствиями. 97 Около половины пациентов умирают в возрасте до 4 лет в результате интеркуррентных заболеваний.
Однако в большинстве случаев болезнь продолжает прогрессировать поэтапно с чередой острых энцефалопатических кризов, вызванных инфекционными заболеваниями, вакцинацией и хирургическим вмешательством в младенчестве или детстве. Часто с возрастом дистония имеет тенденцию эволюционировать от мобильной к стационарной и ассоциироваться с паркинсонизмом. 96 В других случаях он остается неподвижным с тяжелыми двигательными и языковыми последствиями. 97 Около половины пациентов умирают в возрасте до 4 лет в результате интеркуррентных заболеваний.
Его патогенез не ясен. Концентрации глутаровой и 3-гидроксиглутаровой кислоты увеличиваются не только в головном мозге, но и во всех тканях. 3-гидроксиглутаровая и глутаровая кислоты имеют структурное сходство с глутаматом основной возбуждающей аминокислоты, и считается, что повышенные уровни этих кислот играют важную роль в патофизиологии заболевания.3-гидроксиглутаровая кислота вызывает эксайтотоксическое повреждение клеток. Кроме того, глутаровая и 3-гидроксиглутаровая кислоты косвенно модулируют глутаматергическую и ГАМКергическую нейротрансмиссию (что приводит к низким уровням ГАМК), что приводит к дисбалансу возбуждающей и тормозящей нейротрансмиссии. 88
Спектр Леша – Найхана
Спектр Леша – Найхана представляет собой Х-сцепленное нарушение метаболизма пуринов, возникающее в результате дефицита фермента гипоксантин-гуанинфосфорибозилтрансферазы (HGPRT), расположенного на Xq26. 98 , 99 Возможна пренатальная диагностика.
Существует непрерывный спектр неврологических проявлений в зависимости от степени ферментативной недостаточности. Наиболее тяжелые формы известны как болезнь Леша – Нихана (ЛБД). У пациентов с частичным дефицитом HPRT симптомы различаются по интенсивности. В наименее тяжелой форме, называемой синдромом Келли-Сигмиллера, у пациентов наблюдается подагра без неврологических нарушений. 100 Термин «варианты Lesch-Nyhan» был введен для обозначения пациентов с подагрой, связанной с HPRT, и некоторой степенью неврологического поражения, но без полной симптоматики LND.
LND обычно начинается в возрасте от 6 до 18 месяцев с задержкой психомоторного развития, гипотонией или спастичностью. Ранним признаком может быть появление в подгузниках кристаллического вещества оранжевого цвета. Аномальные движения начинаются с тонких атетоидных движений кистей и стоп. В полностью развитом состоянии движения преимущественно дистонические, но могут включать хорею и тремор. На моторику глаз влияют саккады, которым предшествуют движения головы. 101 Примечательной особенностью является агрессивное поведение (85% случаев), направленное как на самого пациента, так и на окружающих.Автоматизация, особенно поражающая губы и пальцы, обычно начинается с прорезывания зубов. Дизартрия также является частым симптомом. Сообщалось об эпилепсии, атрофии зрительного нерва и рецидивирующей коме. 102 , 103 У больных детей также наблюдаются экстраневрологические симптомы, такие как гематурия, кристаллурия и признаки гиперурикемии, такие как почечные камни, подагрический артрит и тофусы. Другие особенности включают рвоту, макроцитарную анемию, замедленный рост и задержку костного возраста.Сейчас часты случаи, когда они выживают после третьего десятилетия жизни.
В вариантах Леша – Нихана степень дистонии менее тяжелая и проявляется в виде дистонической походки, затруднений речи и спастичности, но с нормальными когнитивными функциями и поведением (без членовредительства).
Патофизиология неврологических и поведенческих нарушений остается неясной. Предполагается, что неврологические симптомы не являются результатом избыточного производства мочевой кислоты. Патологоанатомические исследования не выявили каких-либо структурных изменений, но выявили потерю дофамина в базальных ганглиях на 60–90%. 104 Биохимические исследования спинномозговой жидкости и ПЭТ также показали снижение уровня метаболитов дофамина, тогда как серотонин и 5-гидроксииндолуксусную кислоту были увеличены. 105
Гомоцистинурия
Гомоцистинурия вызывается различными ферментативными дефектами. Чаще всего встречается дефицит цистатионин-β-синтазы. Сообщалось о нескольких случаях гомоцистинурии с медленно прогрессирующей дистонией как поздним симптомом. 106 , 107 В этих случаях возраст начала дистонии колеблется от 4 до 21 года.
Причина двигательного расстройства не выяснена. Вероятно сосудистое заболевание. При гомоцистинурии сообщалось об артериальных и венозных тромбозах коры головного мозга и базальных ганглиев, но не было описано нейровизуализационных доказательств поражения базальных ганглиев в случаях дистонии. Кроме того, при патологоанатомическом исследовании сосудов базальных ганглиев патологии не выявлено. 108 , 109
Болезнь Хартнупа
Болезнь Хартнупа является наследственным рецессивным дефектом транспорта аминокислот из-за мутаций в гене SLC6A19 . 110 Светочувствительный дерматит обычно является первым симптомом, возникающим в позднем младенческом или юношеском возрасте. Рецидивирующие эпизоды атаксии — частый неврологический симптом. Сообщалось о дистонических особенностях и перемежающейся дистонии. 111 Возможно самопроизвольное улучшение. Терапия никотинамидом может улучшить неврологические симптомы.
Болезнь Ниманна – Пика, тип C (NPC)
Болезнь Ниманна – Пика, тип C — аутосомно-рецессивное нарушение накопления липидов, которое характеризуется нарушенным внутриклеточным гомеостазом холестерина и дефектным перемещением холестерина через позднюю эндосомальную / лизосомную систему. 112 Возникающее в результате накопление неэтерифицированного холестерина, преимущественно в клетках селезенки, печени и мозга, приводит к ряду клинических проявлений. 113 К ним относятся гепатоспленомегалия, психические расстройства и неврологические проблемы. Мутировавший ген ( NPC1 ) на хромосоме 18q11–12 отвечает за более чем 90% случаев. 109 Несколько случаев связаны с мутациями в гене HE1 / NPC2 . 114
Заболевание проявляется вертикальным надъядерным параличом взора, особенно при взгляде вниз.Клинические проявления в начале обычно включают снижение интеллекта, дизартрию и нарушение походки. Двигательное расстройство может преобладать и состоит в основном из дистонии. Затем появляются мозжечковые и пирамидные признаки. Эпилепсия встречается примерно у трети пациентов. Органомегалия встречается часто (но не постоянно). 115 Магнитно-резонансная томография (МРТ) головного мозга показывает диапазон от нормальной до легкой задней перивентрикулярной гиперинтенсивности белого вещества.
Анализ костного мозга показывает пенистые клетки или гистиоциты цвета морской волны.NPC можно диагностировать с помощью культуры фибробластов кожи, демонстрируя недостаточную этерификацию холестерина, производного от липопротеинов низкой плотности, или окрашивая свободный внутриклеточный холестерин на филиппин.
Нет специального лечения для NPC. Миглустат, иминосахар, ингибирующий глюкозилцерамидсинтазу, может показать клиническую пользу. 116
Другие неврологические расстройства с дистонией
Инфантильный двусторонний стриатальный некроз, детский таламический некроз
Friede (1975) предложил термин «детский двусторонний стриарный некроз» (IBSN) для обозначения невропатологического синдрома с двусторонней симметричной губкой путамина и хвостатые ядра, а иногда и таламуса. 117 Число случаев увеличилось с переходом от невропатологического к нейровизуализационному синдрому. Поражения теперь можно распознать у живых пациентов с помощью компьютерной томографии (КТ), которая показывает гиподенсированные изображения в пораженных областях 118 и, что лучше, с помощью МРТ, которая показывает повышенный сигнал Т2 в тех же зонах. 119
Прогресс в знаниях о митохондриальных нарушениях и органических ацидуриях показывает, что симметричные стриарные поражения являются обычным явлением при этих заболеваниях, и предполагает, что многие из случаев, зарегистрированных как IBSN, могли быть вызваны метаболическими нарушениями, декомпенсированными острой инфекцией, например, глутаровая ацидурия. 120 Следовательно, те случаи, в которых не было выявлено никакой органической ацидурии и не был зарегистрирован лактоацидоз, вероятно, являются недоказанными митохондриальными нарушениями.
Типичные случаи острого СРКН характеризуются острым началом ригидности, дистонией, хореей, баллизмом и стереотипной реакцией на раздражители, приятные или болезненные, с монотонным плачем, гримасой и чрезмерным растяжением шеи. 121 Эпилепсия встречается редко. У некоторых пациентов симптомы развиваются вскоре после различных инфекций, включая инфекции дыхательных путей и эпидемический паротит, вызванный Mycoplasma pneumoniae. 122 На снимках часто наблюдается обширный отек белого вещества в дополнение к симметричной гиподности полосатого тела. Поражения при МРТ могут исчезнуть или сохраниться со временем. 123 Некоторые пациенты могут умереть вскоре после острой фазы, но обычно наступает постепенное улучшение, в некоторых случаях даже через несколько месяцев. Могут возникнуть такие последствия, как дистония или гемипарез. 118 , 123
Хотя некоторые семейные случаи зарегистрированы, большинство случаев IBSN носят спорадический характер. 124 Патогенез остается неизвестным, но возможен параинфекционный механизм. Сообщалось о случаях острого IBSN, представленного вскоре после стрептоккального фарингита, с высокими титрами стрептококка и антителами, реактивными против компонентов базальных ганглиев, что предполагает аутоиммунную этиологию в этих случаях. 125
Инфантильный таламический некроз
Случаи острого некроза обоих таламусов («острая некротическая энцефалопатия у детей») после острой вирусной инфекции первоначально были зарегистрированы из Японии, где это состояние, по-видимому, преобладает. 126 Сообщалось также о кавказцах. 127 Половина случаев остро началась в возрасте от 6 до 18 месяцев. 126 Гипертермия, кома, судороги и децеребрационные или декортикальные позы обычны в острой стадии. Течение часто бывает тяжелым, приводя к смерти в четверти случаев или к серьезным остаточным нарушениям с квадриплегией, микроцефалией, дистонией и умственной отсталостью, но существует «легкая» форма с избирательным обратимым поражением таламуса и полным выздоровлением. 128 В начале курса двусторонние гиперэхогенные таламические поражения могут наблюдаться с помощью ультразвука. КТ или МРТ покажут мультифокальные симметричные поражения в таламусе, а также обычно выявляют поражения в мосту, среднем мозге, мозжечке и белом веществе. Может присутствовать гиперпротеиноррахия во время острого заболевания. 129 Обычно присутствуют повышенные сывороточные CPK, GOT, GPT и лактатдегидрогеназа, но они редко имеют гипераммониемию или гипогликемию, что важно для дифференциации диагноза синдрома Рейе. 126 Причинные или ассоциированные вирусные инфекции, обычно грипп A, были продемонстрированы в нескольких случаях. 129 , 130 Прогноз лучше у детей старшего возраста и пациентов с низкими значениями GOT, без повреждений ствола головного мозга и нормализации нейровизуализации при последующем наблюдении. 128 Сообщалось об эффективном лечении стероидами. 131 Эти случаи показывают некоторое сходство с таковыми из ISBN, поэтому был предложен термин «детский таламический двусторонний некроз». 127
Биотин-зависимая болезнь базальных ганглиев
Биотин-зависимая болезнь базальных ганглиев вызывается дефицитом переносчика тиамина-2 (hTHTR2) из-за мутаций в гене SLC19A3 . 132 Биотин-зависимая болезнь базальных ганглиев (BBGD) была впервые описана в 1998 году у 10 арабских пациентов, за которой последовали новые генетически подтвержденные случаи, состоящие из подострых энцефалопатических эпизодов, характеризующихся спутанностью сознания, рвотой с последующими судорогами, потерей речи или дизартрией, параличом взгляда и т. Д. дисфагия, дистония и общая скованность, которые в конечном итоге привели к коме и смерти. 133 , 134 Введение высоких доз биотина приводит к частичному или полному улучшению в течение нескольких дней. При отсутствии лечения заболевание может привести к прогрессирующей энцефалопатии. Характерная МРТ головного мозга в основном показывает двусторонний некроз хвостатых ядер и скорлупы и, если они есть, незначительные изменения в таламусе. Сообщалось также о пользе тиамина.
Чередующаяся гемиплегия в детском возрасте
Чередующаяся гемиплегия в детском возрасте (AHC) — это заболевание, проявляющееся в возрасте до 18 месяцев эпизодами гемиплегии, поражающими обе стороны и исчезающими (иногда всего на несколько минут) во время сна.Продолжительность эпизодов может составлять от нескольких минут до нескольких дней. Характерными признаками являются эпизоды смещения или двустороннего паралича, связанного со косоглазием или односторонним нистагмом. Часто наблюдаются задержка развития и эпилепсия. Причина неизвестна, лабораторных или нейровизуализационных маркеров нет. Заболевание считалось формой мигрени, хотя эта гипотеза оспаривается.
При AHC тонические и дистонические эпизоды, которые часто носят односторонний характер, присутствуют в 90% случаев и часто являются первым проявлением. 130 Двигательное расстройство, состоящее из дистонии, миоклонии или хореи, первоначально появляется только во время эпизодов, но имеет тенденцию становиться постоянным. Генерализованная, но асимметричная дистония или хореоатетоз почти постоянны на поздних стадиях. 135 , 136 Флунаризин частично эффективен примерно у половины пациентов. 137
Варианты лечения у детей
Лечение большинства форм дистонии симптоматическое, и его польза может быть неполной или сопровождаться невыносимыми побочными эффектами.Эти варианты включают лекарства (системные или очаговые методы лечения, такие как ботулотоксин) и хирургические вмешательства. Существует несколько методов лечения, включая антихолинергические препараты, дофаминовые блокаторы и истощающие препараты, баклофен и бензодиазепины, которые были рекомендованы на основе небольших неконтролируемых исследований. 138
Среди медицинских вариантов обычно первым выбирают высокие дозы холинолитиков. Однако абсолютная и сравнительная эффективность и переносимость антихолинергических средств при дистонии у детей плохо документирована.Таким образом, нельзя давать никаких рекомендаций по назначению лекарств (точка хорошей практики). 139 Более того, дети обычно переносят более высокие дозы, чем взрослые. Контролируемых исследований эффектов этого типа лечения не проводилось. Отсутствуют доказательства положительного эффекта тетрабеназина, чтобы дать рекомендации по этому типу лечения (точка надлежащей практики). Карбидопа / леводопа — это основа лечения в РРБ. Однако другие формы дистонии могут частично поддаваться лечению леводопой.Поскольку полный ответ на леводопу может быть диагностическим для DRD и из-за его потенциальной пользы, всем детям с дистонией настоятельно рекомендуется испытание карбидопы / леводопы. 140
Лечение ботулиническим токсином — это лечение первого выбора при большинстве типов очаговой дистонии. У детей он используется реже, потому что дистонические формы в основном являются генерализованными, но он также может быть полезен для контроля наиболее инвалидизирующих симптомов сегментарной или генерализованной дистонии. 141 , 142 Ботулинический токсин может рассматриваться как лечение первой линии при первичной краниальной (за исключением оромандибулярной) или шейной дистонии.Ботулинический токсин безопасен и эффективен при повторных курсах лечения в течение многих лет, но чрезмерные кумулятивные дозы могут быть опасными, особенно у детей. После предварительных предположений о том, что интратекальное введение баклофена может улучшить дистонию, последующие наблюдения показали, что эта форма лечения особенно полезна, когда спастичность сосуществует с дистонией, например, при церебральном параличе. 143 — 146
Долгосрочная электростимуляция внутреннего бледного шара (GPi) в настоящее время признана эффективным средством лечения различных типов двигательных нарушений, включая дистонию.Использование DBS при дистонии в настоящее время касается, в частности, первичных генерализованных или сегментарных форм у пациентов, которые не достигают достаточного облегчения при консервативных подходах. Паллидный DBS считается хорошим вариантом, особенно при первичной генерализованной или сегментарной дистонии, после того, как лекарства или ботулотоксин не привели к адекватному улучшению. Было замечено, что пациенты с дистонией DYT1 имеют лучший результат, чем пациенты с отрицательным результатом DYT1 , и что чем раньше будет проведено вмешательство в течение болезни, тем лучше результат. 147 , 148 DBS особенно эффективен у детей, страдающих генерализованной дистонией. Невозможно показать, что возраст пациента имеет прямое прогностическое значение для результата. 148 — 150 Более короткая продолжительность болезни — независимо от ее начала — усиливает эффект стимуляции. Эти данные поддерживают раннее вмешательство. Таким образом, при ранней дистонии у детей не следует откладывать возрастное вмешательство.Предварительная информация о небольшом количестве пациентов с M-D, получавших паллидальный DBS, указывает на то, что как миоклонические признаки, так и дистония могут улучшиться. 151
В обзоре 109 пациентов с наследственно-дегенеративными и вторичными синдромами, получавших имплантаты GPi-DBS, некоторые случаи с благоприятными исходами были зарегистрированы почти во всех группах. 152 Кроме того, у восьми из 13 пациентов с PKAN отмечалось резкое улучшение моторики. 153
Обновленная информация о детских дистониях: этиология, эпидемиология и лечение
Реферат
Дистония — это двигательное расстройство, характеризующееся устойчивыми мышечными сокращениями, вызывающими скручивание, повторяющиеся и шаблонные движения или неправильные позы.Дистония — одно из наиболее часто наблюдаемых двигательных расстройств в клинической практике как у взрослых, так и у детей. Он классифицируется на основе этиологии, возраста появления симптомов и распределения пораженных участков тела.
Этиология
Этиология детской дистонии весьма неоднородна. Существует множество различных генетических синдромов и несколько причин симптоматических синдромов. Дистония может быть вторичной по отношению практически к любому патологическому процессу, поражающему двигательную систему, и особенно базальные ганглии.
Классификация
Этиологическая классификация различает первичную дистонию без идентифицированной экзогенной причины или признаков нейродегенерации и вторичных синдромов.
Лечение
Лечение большинства форм дистонии симптоматическое и включает в себя лекарственные препараты (системные или очаговые методы лечения, такие как ботулинический токсин) и хирургические процедуры. Есть несколько лекарств, в том числе антихолинергические, дофамин-блокирующие и истощающие агенты, баклофен и бензодиазепины.У пациентов с нарушениями синтеза дофамина лечение L-допа может быть очень полезным. Лечение ботулиническим токсином может быть полезным в борьбе с наиболее ограничивающими симптомами сегментарной или фокальной дистонии. Длительная электрическая стимуляция внутреннего бледного шара оказывается особенно успешной у детей, страдающих генерализованной дистонией.
Ключевые слова: двигательные расстройства, детская дистония, первичные дистонии, вторичные дистонии
Введение
Дистония — это двигательное расстройство, характеризующееся устойчивыми мышечными сокращениями, вызывающими скручивание, повторяющиеся и шаблонные движения или аномальные позы.Дистония имеет широкий клинический спектр: от минимальных или доброкачественных самоограничивающихся признаков до тяжелых случаев. 1 — 3 Дистония классифицируется на основе: (i) этиологии, (ii) возраста появления симптомов и (iii) распределения пораженных участков тела. Этиология детской дистонии неоднородна (). 4 Этиологическая классификация различает первичную дистонию без идентифицированной экзогенной причины или признаков нейродегенерации и вторичных синдромов.Первичные заболевания можно разделить на чистые, плюс и пароксизмальные. В первичных чистых формах дистония является единственным признаком заболевания (за исключением тремора), а причина либо генетическая, либо неизвестная. Первичные плюсовые синдромы характеризуются дистонией, связанной с дополнительным двигательным расстройством (например, миоклонусом или паркинсонизмом). Наконец, первичные пароксизмальные синдромы включают состояния, характеризующиеся аномальными движениями, включая дистонию, возникающую в виде коротких эпизодов с нормальным состоянием между ними.Вторичные дистонические формы включают синдромы, при которых дистония является ярким признаком наследственно-дегенеративного состояния или когда дистония вызвана экзогенными факторами (например, перинатальная травма, лекарства, опухоль головного мозга, инфекции).
Таблица 1
Классификация дистоний по этиологии
| Первичная | |
|---|---|
| • Первичная чистая | Дистония является единственным клиническим признаком (кроме тремора), и нет никакой идентифицируемой экзогенной причины или другое наследственное или дегенеративное заболевание |
| • Первичная плюс | Дистония является заметным признаком, но связана с другим двигательным расстройством, например миоклонусом или паркинсонизмом.Нет свидетельств нейродегенерации. |
| • Первичная пароксизмальная | Дистония возникает короткими эпизодами с нормальным состоянием между ними. Эти расстройства классифицируются как идиопатические (часто семейные, хотя встречаются и спорадические случаи) и симптоматические из-за множества причин |
| Вторичные | Дистония является симптомом определенного неврологического состояния, такого как очаговое поражение мозга, воздействие лекарств. или химические вещества |
| Наследственная дегенеративная | Дистония — это особенность, среди других неврологических признаков, наследственно-дегенеративного или метаболического расстройства |
Вторая ось классификации различает начало у детей (<18 лет) и начало у взрослых (> 19 лет) кейсы ().Наконец, третья ось, основанная на соматическом распределении дистонии, различает фокальные, сегментарные, многоочаговые, генерализованные и односторонние (гемидистония) формы ().
Таблица 2
Классификация дистоний по возрасту начала
| Раннее начало (варьируется от 20 до 30 лет) | Обычно начинается с конечности и часто распространяется на другие конечности и туловище |
| Позднее начало | Обычно начинается с шеи (включая гортань), черепных мышц или одной руки.Имеет тенденцию оставаться локализованной с ограниченным прогрессированием в соседние мышцы |
Таблица 3
Классификация дистоний по распределению
| Очаговая | Отдельная область тела |
| Сегментарная | Смежные области тела |
| Многоочаговая | Несмежные области тела |
| Обобщенный | Обе ноги и хотя бы одна другая часть тела |
| Гемидистония | Половина тела |
Дистонические синдромы относятся к наиболее часто наблюдаемым двигательным расстройствам в клинической практике как у взрослых, так и у детей, с распространенностью от 2 до 50 случаев на миллион для дистонии с ранним началом (<26 лет) и от 30 до 7320 случаев на миллион для дистонии с поздним началом (> 26 лет). 5 — 7
В этом обзоре мы сосредоточимся на дистонии, которая поражает детей, произвольно ограничиваясь лицами моложе 18 лет. Мы обсудим первичные дистонии с педиатрическим началом, разделенные на «чистые» дистонии (DYT1, DYT6, DYT13, DYT17), «дистонии плюс», связанные с миоклонусом (DYT11 и DYT15), и «дистонии плюс», связанные с паркинсонизмом (DYT5). , DYT12, DYT16). Наконец, мы включим наиболее частые вторичные дистонии.
Синдромы первичной чистой дистонии
В чистом виде дистония является единственным клиническим признаком (кроме тремора).Детская первичная чистая дистония — редкое заболевание; сюда входят семейные и спорадические случаи. Было картировано семь локусов для первичной дистонии, начинающейся у детей, включая DYT1, DYT4, DYT6, DYT13 и DYT17 (). Среди них на данный момент известны только два причинных гена, DYT1 / TOR1A и DYT6 / THAP1. 8
Таблица 4
Молекулярная классификация непароксизмальных дистоний, начинающихся у детей
| Ген | Локус | Обозначение | Продукт гена | Тип |
|---|---|---|---|---|
| С аутосомно-доминантным наследованием | ||||
| DYT1 | 9q34 | Идиопатическая торсионная дистония (ITD) | Торсин A ( TOR1A ) | Первичный чистый |
| DYT4 | Неизвестно | Идиопатическая торсионная дистония | Неизвестная торсионная дистония Неизвестная торсионная дистония 4949||
| DYT5 | 14q22.1 | Дефицит GTPCH | GTP-циклогидролаза 1 ( GCh2 ) | Первичный плюс |
| DYT6 | 8p11–21 | Подростковое / раннее взрослое начало | N-концевой домен THAP ( THAP1 ) | Первичный чистый |
| DYT11 | 7q21 AD | Миоклоническая дистония | ε-саркогликан ( SGCE ) | Первичный плюс |
| DYT12 | 9q13 AD | Rapid-onset + / K + -ATPase ( ATP1A3 ) | Основной плюс | |
| DYT13 | 1p36.13–36,32 | Дистония с ранним началом с поражением шейного отдела черепа и верхних конечностей | Неизвестно | Первичная чистая |
| DYT15 | 18p11 | Миоклоническая дистония, чувствительная к алкоголю | Неизвестно | |
| DYT16 | 2q31.2 | Дистония-паркинсонизм с молодым началом | Киназа PKR ( PRKRA ) | Первичная плюс |
| DYT17 | 20p11.22-q13.12 | Аутосомно-рецессивный первичный очаговый TD | Неизвестно | Первичный чистый |
| С Х-сцепленным наследованием | ||||
| DYT3 | Xq13.1 | Х-сцепленная дистония / паркинсонизм | Фактор 1, связанный с TATA-связывающим белком ( TAF1 ) | Первичный плюс |
| Xq13 | X-связанный синдром дистонии-глухоты (синдром Мора-Транебьерга) | Импортная транслоказа внутренней мембраны митокондрий Tim8A ( TIMM8A ) | ||
DYT1 (
TOR1A ): Дистония с ранним началомДистония с ранним началом, вызванная мутацией DYT1 , представляет собой наиболее частую и наиболее тяжелую форму первичной дистонии.На его долю приходится 16–53% случаев детской дистонии (POD) среди нееврейского населения и 80–90% пациентов среди еврейского населения ашкенази. 9 — 11
Рассчитанная частота заболевания среди еврейского населения ашкенази составила 1: 3000–1: 9000. Среди нееврейского населения частота примерно в пять раз ниже. 12 , 13
Обычно дистония DYT1 начинается в детстве или подростковом возрасте. Первоначальным симптомом обычно является дистония очагового действия, затрагивающая одну конечность (писчая дистония, ходячая дистония с инверсией или выворотом стопы).Дистония впоследствии распространяется на другие области тела, становится менее специфичной и может также присутствовать в состоянии покоя. Пациенты с дистонией ног в начале болезни обычно имеют более раннее начало (средний возраст 9 лет) и генерализованную дистонию через несколько месяцев или лет. Пациенты с дистонией руки имеют более позднее начало (средний возраст 15 лет), и генерализация дистонии встречается реже. Дистония также может начаться в шее или черепных мышцах. 14
В целом, до 65% пациентов с дистонией DYT1 имеют генерализованную дистонию, при которой чаще всего поражаются конечности.Черепные мышцы поражаются реже, примерно у 11–18% пациентов. Пораженные члены семьи могут иметь начало у взрослых и чаще с поражением шеи. Сообщалось о необычных фенотипах, таких как изолированный блефароспазм и односторонняя миоклоническая дистония. 15
У некоторых пациентов с дистонией DYT 1 может наблюдаться резкое ухудшение дистонии, называемое «status dystonicus», в течение болезни. 16 Продукт гена DYT1 , TorsinA, является членом суперсемейства АТФаз AAA1, связанных с различными клеточными активностями.Он выполняет важные функции, связанные с деградацией белков, переносом мембран, слиянием везикул и перемещением органелл, динамикой цитоскелета и правильным сворачиванием белков. 17
TorsinA экспрессируется почти повсеместно, и его экспрессия в головном мозге ограничена нейронами, где он связан с эндоплазматическим ретикулумом (ER). В клеточных моделях, экспрессирующих патогенную делецию GAG, мутантный TorsinA перераспределяется из просвета ER в ядерную оболочку (NE). 18 Эти клетки также обнаруживают аномальную морфологию и утолщение NE, включая образование включений мутовидной мембраны, которые, по-видимому, происходят из ER и NE. Эти включения связаны с везикулярным транспортером моноаминов VMAT2, открытие, которое может функционально связать TorsinA с дофаминергической системой. 19 Кроме того, было обнаружено, что TorsinA регулирует клеточный перенос переносчика дофамина и других мембраносвязанных белков. Также было показано, что мутант TorsinA вмешивается в процессы цитоскелета, которые могут влиять на развитие нейрональных путей в головном мозге, и что он преждевременно разрушается как протеасомными путями, так и путями макроаутофагии. 20 Недавно было показано, что TorsinA контролирует стабильность белков синаптических везикул и влияет на оборот синаптических везикул в нейронах. 21
Первичные дистонии с ранним началом, не связанные с DYT1
Большая группа пациентов с ПОЛ не связана с мутацией DYT1 . DYT6 дистония была описана в двух семьях германо-меннонитского происхождения. 22 Фенотип определяется как смешанный, потому что он отличается от типичного педиатрического начала: варьирующий возраст в начале от младенчества до взрослого возраста (диапазон 5–38 лет; среднее значение 18.6 лет), выраженное поражение черепных и шейных мышц, генерализация дистонии наблюдается примерно у половины пациентов. 23 Продукт гена DYT6 , Танатос-ассоциированный белок 1 (THAP1), является членом семейства клеточных факторов, разделяющих высококонсервативный ДНК-связывающий домен THAP1, который представляет собой атипичный цинковый палец и может регулировать эндотелиальные клетки. распространение. 24 Предполагаемый механизм заболевания заключается в том, что мутации DYT6 могут нарушать связывание THAP1 с ДНК и вызывать нарушение регуляции транскрипции.Недавно было продемонстрировано, что между THAP1 и промотором TOR1A происходит физическое взаимодействие, которое устраняется патогенными мутациями. 25
Эта связь, вероятно, не просто простое взаимодействие промоторов THAP1-Tor1A, но, вероятно, также включает другие гены. Интересно, что THAP1 также связывается с промоторной областью транскрипта TAF1, который участвует в форме первичной плюс дистонии (Х-сцепленная паркинсонизм с началом Х-хромосомы). 26
Другой редкой причиной раннего начала первичной дистонии является мутация DYT13 , которая была обнаружена в большой итальянской семье. 27 Преобладающим фенотипом является детская дистония верхней части тела с частым поражением черепно-шейной области и относительно доброкачественным течением.
Первичная дистония DYT17 описана как пример аутосомно-рецессивного POD. 28 , 29 Локус для DYT17 был назначен на хромосому 20.
Первичные плюсовые синдромы
Дистония является заметным признаком дистонии плюс синдромов, но она связана с другими двигательными расстройствами (например, myoclonus или паркинсонизм) без признаков нейродегенерации.Выделяют три клинически определяемых состояния: допа-зависимая дистония ( DRD; DYT5 ), миоклоническая дистония (MD; DYT11 ) и быстро развивающаяся дистония-паркинсонизм (RDP; DYT12 ), которые характеризуются выраженным фенотипическая гетерогенность.
DRD или болезнь Сегавы
DRD или болезнь Сегавы в основном возникает у детей из-за дефектов синтеза дофамина. Обычный возраст начала заболевания колеблется от 4 до 6 лет. Соотношение самок и самцов — 2.5: 1. 30 Сообщалось о распространенности от 0,5 до 1 на миллион, но, вероятно, она недооценена, поскольку пенетрантность низкая, а атипичные случаи часты. 31 Начало часто коварное, с утомляемостью, неуклюжей походкой и дистоническими позами, часто ограниченными одной стопой. 32 , 33
У детей старше 10 лет первыми проявлениями могут быть дистония верхних конечностей или постуральный тремор. 34 Обычно присутствуют легкие признаки паркинсонизма, такие как ригидность конечностей и туловища и гипомимия.Очень характерно прогрессирующее усиление дистонии в течение дня и заметное уменьшение, а иногда и полное исчезновение после сна. Расстройство прогрессирует и может привести к тяжелой инвалидности. Поражение других частей тела может произойти быстро или занять до десяти лет. Когнитивные функции сохранены.
Дистония и / или паркинсонизм обычно показывают быстрый, выраженный и устойчивый ответ на низкие дозы (3-5 мг / кг / день) L-допа в сочетании с ингибитором периферического декарбоксилирования, помогающим установить клинический диагноз.Ответ не зависит от задержки в начале лечения. Дефицит аутосомно-доминантной гуанозинтрифосфатциклогидролазы (GTPCH) ( DYT5 ) является наиболее частым ферментативным дефектом, приводящим к болезни Сегавы. Почти 75% случаев индекса DRD большой европейской серии показали этот дефект. 35
Сообщалось об аутосомно-рецессивных формах GTPCH. 36 Аутосомно-рецессивные формы DRD (ArDRD) также возникают в результате гомозиготных или сложных гетерозиготных мутаций тирозингидроксилазы (TH) и сепиаптеринредуктазы (SPR). 37 При ArDRD фенотип обычно более тяжелый, включая когнитивные нарушения и задержку развития, и его также можно назвать инфантильным энцефалопатическим фенотипом (IEP). 3 Ответ на лечение L-допа при IEP отличается от болезни Сегавы. Только в редких случаях низкие дозы L-допы могут привести к быстрому улучшению симптомов. У большинства этих пациентов низкие дозы L-допы могут вызывать выраженные хореические и / или дистонические движения. У большинства этих пациентов необходимо очень медленное титрование дозы, и часто невозможно достичь эффективных терапевтических уровней L-допа.Даже низкие дозы могут вызывать заметные хореические и / или дистонические движения. Прогноз ИЭП отличается от прогноза болезни Сегавы даже в тех случаях, когда частично отвечает дофаминергическая терапия; Часто сообщается о неуклюжести и легкой умственной отсталости.
Недавно у некоторых пациентов с ИЭП, вызванным мутациями в гене, кодирующем переносчик допамина ( SLC6A3 ), было зарегистрировано новое состояние, называемое синдромом дефицита переносчика дофамина (DTDS). Исследования нейротрансмиттеров спинномозговой жидкости показали повышенное соотношение HVA / HIAA.Сканирование переносчика дофамина с помощью однофотонной эмиссионной компьютерной томографии (DaTSCAN SPECT) показывает полную потерю активности переносчика дофамина в базальных ядрах. 38
Миоклоническая дистония (M-D) (
DYT11 )Заболевание начинается в детском или подростковом возрасте. Симптомом обычно является миоклонус, поражающий верхнюю часть тела (шею, туловище, конечность) с преобладанием проксимальных мышц. Также могут быть затронуты черепные мышцы, в том числе мышцы гортани. 39 — 43 Дистония связана с миоклонусом более чем у половины пациентов и обычно вызывает незначительную инвалидность или не вызывает ее вовсе. Дистония обычно поражает шею или руки (кривошея, судорога писателя). 40 , 41 , 44 — 46 Заметное улучшение двигательных симптомов (в основном миоклонуса) после приема алкоголя и наличия психических симптомов, таких как обсессивно-компульсивные расстройства, злоупотребление алкоголем часто сообщается о депрессии и тревоге. 44 , 47 — 49 Течение болезни, как правило, доброкачественное, но исходы у пациентов, даже в пределах одной семьи, сильно различаются. 41
M-D вызывается гетерозиготными мутациями гена эпсилон-саркогликана ( SGCE ). Выявлено более 50 различных гетерозиготных мутаций SGCE . 50 Считается, что эти мутации вызывают потерю функции и не позволяют провести корреляцию генотип-фенотип.Однако заметные различия в степени тяжести заболевания предполагают влияние факторов окружающей среды или генетических модификаторов. Геномной особенностью SGCE является материнский геномный импринтинг. 51
Дистония-паркинсонизм с быстрым началом (RDP,
DYT12 )Заболевание характеризуется внезапной дистонией и паркинсонизмом. 52 , 53 В своей генетической форме тип наследования аутосомно-доминантный с пониженной пенетрантностью, и сообщалось о мутациях de novo. 54 , 55 Одной из моногенетических причин RDP является мутация в гене альфа-субъединицы Na + / K + -АТФазы 3 (a3; ATP1A3 ). Хотя зарегистрировано всего несколько семей и единичные случаи с этим заболеванием, клинические особенности делают это заболевание особенно интересным. Расстройство обычно начинается в возрасте от 15 до 45 лет. Отличительными признаками являются резкое появление двигательных симптомов в течение от нескольких минут до 30 дней с вовлечением рострокаудальной области и выраженными бульбарными находками.Начало часто следует за физическим или эмоциональным стрессом, приводящим к стойкой неврологической инвалидности. 54 Большинство пациентов остаются стабильными или демонстрируют небольшое улучшение через годы после резкого появления симптомов. Заболевание передается по аутосомно-доминантному признаку с переменной выраженностью и пониженной пенетрантностью. Мутации в гене ATP1A3 (хромосома 19q13), связанном с NA + / K + ATPase alpha3, являются причиной нарушения. 54 , 55 RDP — первое известное дистоническое состояние, связанное с аномалией мембранного ионного канала.Все исследования остаются отрицательными, за исключением низкого уровня HVA в спинномозговой жидкости, что свидетельствует о пресинаптическом дефекте нигростриатных путей. 56
Недавно был идентифицирован новый ген PRKRA (протеинкиназа, интерферон-индуцибельный двухцепочечный РНК-зависимый активатор) для молодой дистонии-паркинсонизма ( DYT16 ). 57
Другие первичные генерализованные дистонии
Х-связанная дистония глухоты
Сообщалось о нескольких семьях с синдромом Х-связанной дистонии-глухоты (также называемым синдромом Мора – Транебьерга). 58 , 59 Заболевание характеризуется началом сенсоневральной глухоты обычно в возрасте до 2 лет. Тяжелая прогрессирующая генерализованная дистония начинается примерно в 7-летнем возрасте. Сообщалось о случаях развития дистонии уже в возрасте 30 лет, что позволяет предположить, что клинические признаки имеют более широкий спектр, чем считалось ранее. 60 , 61 Большинство испытуемых приковывались к инвалидной коляске в возрасте от 9 до 22 лет.Умственная отсталость, нарушение зрения из-за корковой слепоты или связанное с нейропатией зрительного нерва, 62 вовлечение кортикоспинального тракта и психиатрические проявления (раздражительность, беспокойство, паранойя) могут быть ассоциированными признаками. Ген-кандидат для этого расстройства, названный DDP (пептид глухоты / дистонии) или TIMM8A (вероятно, кодирующий импортируемую в митохондрии субъединицу транслоказы внутренней мембраны Tim8 A, цинк-связывающий белок), был идентифицирован на хромосоме Xq22. 63 Белок (белок DDP) расположен в межмембранном пространстве митохондрий, поэтому мутированный белок DDP может нарушить функцию митохондрий. 64 Сообщалось о нескольких мутациях, включая интронные. 65 По крайней мере, в одной из описанных мутаций самки-носители также страдают дистонией, но, в отличие от мужчин, она описывается как очаговая дистония. 66
ГАМКергические вещества, такие как клоназепам и гаммагидроксимасляная кислота, считаются полезными. 60
Преходящая идиопатическая дистония младенчества
Преходящая дистония младенчества обычно проявляется в возрасте до 5 месяцев. 67 , 68 У пораженных младенцев аномальные позы обычно ограничиваются одной верхней или нижней конечностью. Иногда в позы могут вовлекаться туловище, руки или одна сторона тела. 69 В положении лежа младенец часто поддерживает форсированную пронацию предплечья, используя тыльную сторону кисти в качестве опоры. В нижней конечности ступни могут находиться в эквиноварусии. Позы могут быть прерывистыми. 70 В остальном неврологическое обследование и развитие в норме.Двигательное расстройство самопроизвольно исчезает к первому дню рождения. Семейные случаи известны. 67 , 68 Легкие формы могут быть частыми, и только в наиболее выраженных случаях может потребоваться медицинская помощь. Ключом к диагнозу является наблюдение, что дистоническая поза исчезает, когда младенец выполняет целенаправленные движения пораженной конечностью. Этиология пока неизвестна.
При типичных клинических признаках дополнительные исследования (нейровизуализация, лабораторные исследования) не требуются.
Первичные пароксизмальные синдромы
Пароксизмальные дискинезии относятся к приступам аномальных движений и поз (дистонических, хореоатетоидных или их комбинации) с возвращением к нормальному состоянию между эпизодами. 71
Можно выделить три основные категории: пароксизмальная кинезигенная дискинезия (PKD / DYT10), пароксизмальная некинезигенная дискинезия (PNKD1; DYT8) и пароксизмальная дискинезия, вызванная физической нагрузкой (PED; DYT18). 72 В ДОК продолжительность атак варьируется от секунд до часов, а частота эпизодов варьируется от одного в год до сотен в день.PKD характеризуется дистонией или хореодистонией, вызванной внезапной сменой положения, обычно переходящего из положения сидя в положение стоя. PNKD характеризуется спонтанными приступами в покое, которые имеют тенденцию к более дистоническому характеру, хотя могут возникать хорея, атетоз и баллизм. Эпизоды могут длиться от секунд до нескольких часов и иметь длинные интервалы без приступов. Начало в детстве с тенденцией к уменьшению приступов с возрастом. 73 Симптомы могут быть спровоцированы алкоголем или кофеином и, в меньшей степени, никотином, возбуждением, усталостью, голодом и эмоциональным стрессом.Пароксизмальный хореоатетоз с клиническими характеристиками, подобными таковым для PNKD с ассоциированной спастичностью, был описан под термином аутосомно-доминантный пароксизмальный хореоатетоз / синдром спастичности (CSE), и была продемонстрирована связь с хромосомой 1p. 74 В PED приступы возникают после 10 или 15 минут непрерывных упражнений. Приступы обычно дистонические и чаще всего появляются в нижних конечностях после продолжительной ходьбы или бега. Приступы дистонии обычно прекращаются через 10–15 минут после прекращения упражнений. 75
Пациенты с мутациями гена SLC2A1 (ген, кодирующий транспортер Glut 1) могут иметь PED, который может быть связан с эпилепсией и / или умственной отсталостью. 76 — 78
Список локусов сцепленных генов, вызывающих фенотипы пароксизмальной дискинезии, быстро растет, хотя гены большинства этих состояний все еще остаются неидентифицированными. Пароксизмальные дискинезии наследуются по аутосомно-доминантному признаку. 79 PNKD вызывается мутациями в гене PNKD1 , кодирующем регулятор 1 миофибриллогенеза.При PKD локус заболевания был приписан перицентромерной области хромосомы 16 (16p11.2-q12.1). 80 Первый ген, вызывающий PKD, был недавно идентифицирован: трансмембранный белок пролинерих 2. 81 , 82 Ген PED расположен в коротком плече хромосомы 1 (1p31. 3 – p35). Ген SLC2A1 кодирует транспортер глюкозы 1 (Glut1). 77 Поскольку Glut1 доставляет глюкозу через гематоэнцефалический барьер, дискинезии могут быть результатом дефицита энергии в базальных ганглиях при физической нагрузке.
Вторичные дистонии
Дистония часто ассоциируется с другими неврологическими симптомами в длинном списке заболеваний, и затем ее называют вторичной дистонией.
Дистония, вызванная структурным дефектом
Дистонический церебральный паралич и дистония с отсроченным началом не будут обсуждаться в этом обзоре. Гемидистония часто возникает из-за структурного поражения. Наиболее частой причиной гемидистонии является острая окклюзия сосудов любой этиологии, особенно поражающая полосатое тело и / или хвостатое тело. 83 Гемидистония чаще встречается у детей, чем у взрослых. 84 Гемидистония преобладает в верхних конечностях и очень часто сопровождается признаками пирамидного тракта. Первым неврологическим проявлением обычно является острый гемипарез. Дистония может появиться во время выздоровления гемипареза, но часто она становится очевидной только через несколько месяцев или даже лет. 84
Часто оказывается плохой ответ на лечение. Наилучшие результаты дают бензодиазепины и антихолинергические препараты.Может быть полезен ботулинический токсин. Хирургия, в основном глубокая стимуляция головного мозга (DBS) в определенных областях внутреннего бледного шара, кажется сейчас лечением выбора. 85
Наследственные дегенеративные синдромы
Наследственные дегенеративные синдромы составляют большую и разнородную группу заболеваний, при которых дистония может возникать с разной степенью тяжести и связана с другими аномалиями, включая умственную отсталость, судороги, атаксию, невропатию, атрофию зрительного нерва и взор. парез.Многие из этих расстройств являются аутосомно-рецессивными из-за наследственных дегенеративных или метаболических заболеваний и возникают в основном в детстве.
Глутаровая ацидурия и другие органические ацидемии с дистонией
Глутаровая ацидурия I типа (GAI), также называемая дефицитом глутарил-КоА-дегидрогеназы, представляет собой аутосомно-рецессивное заболевание, вызываемое мутациями в гене глутарил-КоА-дегидрогеназы ( GCDH), расположенном на хромосоме GCDH. 19п13.2. 86 , 87 GCDH — митохондриальный фермент, который играет ключевую роль в катаболизме лизина, гидроксилизина и триптофана.Сильное снижение или полное отсутствие активности GCDH вызывает накопление 3-гидроксиглутаровой кислоты и глутаровой кислоты в плазме, моче и спинномозговой жидкости, что может вызвать гибель нейронов из-за эксайтотоксичности, а также дисфункции митохондрий. 88
Частота ГАИ оценивается в 1 на 100 000 новорожденных. 89 Клинические проявления обычно появляются в возрасте от 5 до 14 месяцев, но легкие симптомы, такие как небольшая задержка моторики и гипотония, могут наблюдаться в более раннем возрасте.Макроцефалия при рождении или развившаяся позже в младенчестве присутствует примерно в 70% случаев. 90 , 91 В большинстве случаев заболевание начинается внезапно, с очаговыми припадками или генерализованными судорогами, рвотой и заторможенностью или летаргией, обычно после острого инфекционного заболевания. Позже появляются психомоторный регресс и дистонические или хореоатетотические движения. У некоторых пациентов начало незаметно с медленно развивающейся задержкой психомоторного развития, гипотонией и дистоническими позами до 16 лет. 92 — 95
Характерным неврологическим последствием является генерализованная дистония. Также может быть очевидна спастичность. Часто встречаются сложные речевые изменения с сочетанием признаков гиперкинетической дизартрии, анартрии и апраксии. Понимание языка намного лучше, чем выражение, что говорит о том, что познание относительно сохранено.
Течение расстройства непостоянно. ГАИ — излечимое состояние. В идеале лечение следует начинать до появления клинических симптомов.В таких случаях диета в сочетании с пероральным приемом L-карнитина и агрессивным лечением во время острых эпизодов интеркуррентного заболевания может предотвратить развитие клинических признаков или уменьшить их тяжесть. Однако в большинстве случаев болезнь продолжает прогрессировать поэтапно с чередой острых энцефалопатических кризов, вызванных инфекционными заболеваниями, вакцинацией и хирургическим вмешательством в младенчестве или детстве. Часто с возрастом дистония имеет тенденцию эволюционировать от мобильной к стационарной и ассоциироваться с паркинсонизмом. 96 В других случаях он остается неподвижным с тяжелыми двигательными и языковыми последствиями. 97 Около половины пациентов умирают в возрасте до 4 лет в результате интеркуррентных заболеваний.
Его патогенез не ясен. Концентрации глутаровой и 3-гидроксиглутаровой кислоты увеличиваются не только в головном мозге, но и во всех тканях. 3-гидроксиглутаровая и глутаровая кислоты имеют структурное сходство с глутаматом основной возбуждающей аминокислоты, и считается, что повышенные уровни этих кислот играют важную роль в патофизиологии заболевания.3-гидроксиглутаровая кислота вызывает эксайтотоксическое повреждение клеток. Кроме того, глутаровая и 3-гидроксиглутаровая кислоты косвенно модулируют глутаматергическую и ГАМКергическую нейротрансмиссию (что приводит к низким уровням ГАМК), что приводит к дисбалансу возбуждающей и тормозящей нейротрансмиссии. 88
Спектр Леша – Найхана
Спектр Леша – Найхана представляет собой Х-сцепленное нарушение метаболизма пуринов, возникающее в результате дефицита фермента гипоксантин-гуанинфосфорибозилтрансферазы (HGPRT), расположенного на Xq26. 98 , 99 Возможна пренатальная диагностика.
Существует непрерывный спектр неврологических проявлений в зависимости от степени ферментативной недостаточности. Наиболее тяжелые формы известны как болезнь Леша – Нихана (ЛБД). У пациентов с частичным дефицитом HPRT симптомы различаются по интенсивности. В наименее тяжелой форме, называемой синдромом Келли-Сигмиллера, у пациентов наблюдается подагра без неврологических нарушений. 100 Термин «варианты Lesch-Nyhan» был введен для обозначения пациентов с подагрой, связанной с HPRT, и некоторой степенью неврологического поражения, но без полной симптоматики LND.
LND обычно начинается в возрасте от 6 до 18 месяцев с задержкой психомоторного развития, гипотонией или спастичностью. Ранним признаком может быть появление в подгузниках кристаллического вещества оранжевого цвета. Аномальные движения начинаются с тонких атетоидных движений кистей и стоп. В полностью развитом состоянии движения преимущественно дистонические, но могут включать хорею и тремор. На моторику глаз влияют саккады, которым предшествуют движения головы. 101 Примечательной особенностью является агрессивное поведение (85% случаев), направленное как на самого пациента, так и на окружающих.Автоматизация, особенно поражающая губы и пальцы, обычно начинается с прорезывания зубов. Дизартрия также является частым симптомом. Сообщалось об эпилепсии, атрофии зрительного нерва и рецидивирующей коме. 102 , 103 У больных детей также наблюдаются экстраневрологические симптомы, такие как гематурия, кристаллурия и признаки гиперурикемии, такие как почечные камни, подагрический артрит и тофусы. Другие особенности включают рвоту, макроцитарную анемию, замедленный рост и задержку костного возраста.Сейчас часты случаи, когда они выживают после третьего десятилетия жизни.
В вариантах Леша – Нихана степень дистонии менее тяжелая и проявляется в виде дистонической походки, затруднений речи и спастичности, но с нормальными когнитивными функциями и поведением (без членовредительства).
Патофизиология неврологических и поведенческих нарушений остается неясной. Предполагается, что неврологические симптомы не являются результатом избыточного производства мочевой кислоты. Патологоанатомические исследования не выявили каких-либо структурных изменений, но выявили потерю дофамина в базальных ганглиях на 60–90%. 104 Биохимические исследования спинномозговой жидкости и ПЭТ также показали снижение уровня метаболитов дофамина, тогда как серотонин и 5-гидроксииндолуксусную кислоту были увеличены. 105
Гомоцистинурия
Гомоцистинурия вызывается различными ферментативными дефектами. Чаще всего встречается дефицит цистатионин-β-синтазы. Сообщалось о нескольких случаях гомоцистинурии с медленно прогрессирующей дистонией как поздним симптомом. 106 , 107 В этих случаях возраст начала дистонии колеблется от 4 до 21 года.
Причина двигательного расстройства не выяснена. Вероятно сосудистое заболевание. При гомоцистинурии сообщалось об артериальных и венозных тромбозах коры головного мозга и базальных ганглиев, но не было описано нейровизуализационных доказательств поражения базальных ганглиев в случаях дистонии. Кроме того, при патологоанатомическом исследовании сосудов базальных ганглиев патологии не выявлено. 108 , 109
Болезнь Хартнупа
Болезнь Хартнупа является наследственным рецессивным дефектом транспорта аминокислот из-за мутаций в гене SLC6A19 . 110 Светочувствительный дерматит обычно является первым симптомом, возникающим в позднем младенческом или юношеском возрасте. Рецидивирующие эпизоды атаксии — частый неврологический симптом. Сообщалось о дистонических особенностях и перемежающейся дистонии. 111 Возможно самопроизвольное улучшение. Терапия никотинамидом может улучшить неврологические симптомы.
Болезнь Ниманна – Пика, тип C (NPC)
Болезнь Ниманна – Пика, тип C — аутосомно-рецессивное нарушение накопления липидов, которое характеризуется нарушенным внутриклеточным гомеостазом холестерина и дефектным перемещением холестерина через позднюю эндосомальную / лизосомную систему. 112 Возникающее в результате накопление неэтерифицированного холестерина, преимущественно в клетках селезенки, печени и мозга, приводит к ряду клинических проявлений. 113 К ним относятся гепатоспленомегалия, психические расстройства и неврологические проблемы. Мутировавший ген ( NPC1 ) на хромосоме 18q11–12 отвечает за более чем 90% случаев. 109 Несколько случаев связаны с мутациями в гене HE1 / NPC2 . 114
Заболевание проявляется вертикальным надъядерным параличом взора, особенно при взгляде вниз.Клинические проявления в начале обычно включают снижение интеллекта, дизартрию и нарушение походки. Двигательное расстройство может преобладать и состоит в основном из дистонии. Затем появляются мозжечковые и пирамидные признаки. Эпилепсия встречается примерно у трети пациентов. Органомегалия встречается часто (но не постоянно). 115 Магнитно-резонансная томография (МРТ) головного мозга показывает диапазон от нормальной до легкой задней перивентрикулярной гиперинтенсивности белого вещества.
Анализ костного мозга показывает пенистые клетки или гистиоциты цвета морской волны.NPC можно диагностировать с помощью культуры фибробластов кожи, демонстрируя недостаточную этерификацию холестерина, производного от липопротеинов низкой плотности, или окрашивая свободный внутриклеточный холестерин на филиппин.
Нет специального лечения для NPC. Миглустат, иминосахар, ингибирующий глюкозилцерамидсинтазу, может показать клиническую пользу. 116
Другие неврологические расстройства с дистонией
Инфантильный двусторонний стриатальный некроз, детский таламический некроз
Friede (1975) предложил термин «детский двусторонний стриарный некроз» (IBSN) для обозначения невропатологического синдрома с двусторонней симметричной губкой путамина и хвостатые ядра, а иногда и таламуса. 117 Число случаев увеличилось с переходом от невропатологического к нейровизуализационному синдрому. Поражения теперь можно распознать у живых пациентов с помощью компьютерной томографии (КТ), которая показывает гиподенсированные изображения в пораженных областях 118 и, что лучше, с помощью МРТ, которая показывает повышенный сигнал Т2 в тех же зонах. 119
Прогресс в знаниях о митохондриальных нарушениях и органических ацидуриях показывает, что симметричные стриарные поражения являются обычным явлением при этих заболеваниях, и предполагает, что многие из случаев, зарегистрированных как IBSN, могли быть вызваны метаболическими нарушениями, декомпенсированными острой инфекцией, например, глутаровая ацидурия. 120 Следовательно, те случаи, в которых не было выявлено никакой органической ацидурии и не был зарегистрирован лактоацидоз, вероятно, являются недоказанными митохондриальными нарушениями.
Типичные случаи острого СРКН характеризуются острым началом ригидности, дистонией, хореей, баллизмом и стереотипной реакцией на раздражители, приятные или болезненные, с монотонным плачем, гримасой и чрезмерным растяжением шеи. 121 Эпилепсия встречается редко. У некоторых пациентов симптомы развиваются вскоре после различных инфекций, включая инфекции дыхательных путей и эпидемический паротит, вызванный Mycoplasma pneumoniae. 122 На снимках часто наблюдается обширный отек белого вещества в дополнение к симметричной гиподности полосатого тела. Поражения при МРТ могут исчезнуть или сохраниться со временем. 123 Некоторые пациенты могут умереть вскоре после острой фазы, но обычно наступает постепенное улучшение, в некоторых случаях даже через несколько месяцев. Могут возникнуть такие последствия, как дистония или гемипарез. 118 , 123
Хотя некоторые семейные случаи зарегистрированы, большинство случаев IBSN носят спорадический характер. 124 Патогенез остается неизвестным, но возможен параинфекционный механизм. Сообщалось о случаях острого IBSN, представленного вскоре после стрептоккального фарингита, с высокими титрами стрептококка и антителами, реактивными против компонентов базальных ганглиев, что предполагает аутоиммунную этиологию в этих случаях. 125
Инфантильный таламический некроз
Случаи острого некроза обоих таламусов («острая некротическая энцефалопатия у детей») после острой вирусной инфекции первоначально были зарегистрированы из Японии, где это состояние, по-видимому, преобладает. 126 Сообщалось также о кавказцах. 127 Половина случаев остро началась в возрасте от 6 до 18 месяцев. 126 Гипертермия, кома, судороги и децеребрационные или декортикальные позы обычны в острой стадии. Течение часто бывает тяжелым, приводя к смерти в четверти случаев или к серьезным остаточным нарушениям с квадриплегией, микроцефалией, дистонией и умственной отсталостью, но существует «легкая» форма с избирательным обратимым поражением таламуса и полным выздоровлением. 128 В начале курса двусторонние гиперэхогенные таламические поражения могут наблюдаться с помощью ультразвука. КТ или МРТ покажут мультифокальные симметричные поражения в таламусе, а также обычно выявляют поражения в мосту, среднем мозге, мозжечке и белом веществе. Может присутствовать гиперпротеиноррахия во время острого заболевания. 129 Обычно присутствуют повышенные сывороточные CPK, GOT, GPT и лактатдегидрогеназа, но они редко имеют гипераммониемию или гипогликемию, что важно для дифференциации диагноза синдрома Рейе. 126 Причинные или ассоциированные вирусные инфекции, обычно грипп A, были продемонстрированы в нескольких случаях. 129 , 130 Прогноз лучше у детей старшего возраста и пациентов с низкими значениями GOT, без повреждений ствола головного мозга и нормализации нейровизуализации при последующем наблюдении. 128 Сообщалось об эффективном лечении стероидами. 131 Эти случаи показывают некоторое сходство с таковыми из ISBN, поэтому был предложен термин «детский таламический двусторонний некроз». 127
Биотин-зависимая болезнь базальных ганглиев
Биотин-зависимая болезнь базальных ганглиев вызывается дефицитом переносчика тиамина-2 (hTHTR2) из-за мутаций в гене SLC19A3 . 132 Биотин-зависимая болезнь базальных ганглиев (BBGD) была впервые описана в 1998 году у 10 арабских пациентов, за которой последовали новые генетически подтвержденные случаи, состоящие из подострых энцефалопатических эпизодов, характеризующихся спутанностью сознания, рвотой с последующими судорогами, потерей речи или дизартрией, параличом взгляда и т. Д. дисфагия, дистония и общая скованность, которые в конечном итоге привели к коме и смерти. 133 , 134 Введение высоких доз биотина приводит к частичному или полному улучшению в течение нескольких дней. При отсутствии лечения заболевание может привести к прогрессирующей энцефалопатии. Характерная МРТ головного мозга в основном показывает двусторонний некроз хвостатых ядер и скорлупы и, если они есть, незначительные изменения в таламусе. Сообщалось также о пользе тиамина.
Чередующаяся гемиплегия в детском возрасте
Чередующаяся гемиплегия в детском возрасте (AHC) — это заболевание, проявляющееся в возрасте до 18 месяцев эпизодами гемиплегии, поражающими обе стороны и исчезающими (иногда всего на несколько минут) во время сна.Продолжительность эпизодов может составлять от нескольких минут до нескольких дней. Характерными признаками являются эпизоды смещения или двустороннего паралича, связанного со косоглазием или односторонним нистагмом. Часто наблюдаются задержка развития и эпилепсия. Причина неизвестна, лабораторных или нейровизуализационных маркеров нет. Заболевание считалось формой мигрени, хотя эта гипотеза оспаривается.
При AHC тонические и дистонические эпизоды, которые часто носят односторонний характер, присутствуют в 90% случаев и часто являются первым проявлением. 130 Двигательное расстройство, состоящее из дистонии, миоклонии или хореи, первоначально появляется только во время эпизодов, но имеет тенденцию становиться постоянным. Генерализованная, но асимметричная дистония или хореоатетоз почти постоянны на поздних стадиях. 135 , 136 Флунаризин частично эффективен примерно у половины пациентов. 137
Варианты лечения у детей
Лечение большинства форм дистонии симптоматическое, и его польза может быть неполной или сопровождаться невыносимыми побочными эффектами.Эти варианты включают лекарства (системные или очаговые методы лечения, такие как ботулотоксин) и хирургические вмешательства. Существует несколько методов лечения, включая антихолинергические препараты, дофаминовые блокаторы и истощающие препараты, баклофен и бензодиазепины, которые были рекомендованы на основе небольших неконтролируемых исследований. 138
Среди медицинских вариантов обычно первым выбирают высокие дозы холинолитиков. Однако абсолютная и сравнительная эффективность и переносимость антихолинергических средств при дистонии у детей плохо документирована.Таким образом, нельзя давать никаких рекомендаций по назначению лекарств (точка хорошей практики). 139 Более того, дети обычно переносят более высокие дозы, чем взрослые. Контролируемых исследований эффектов этого типа лечения не проводилось. Отсутствуют доказательства положительного эффекта тетрабеназина, чтобы дать рекомендации по этому типу лечения (точка надлежащей практики). Карбидопа / леводопа — это основа лечения в РРБ. Однако другие формы дистонии могут частично поддаваться лечению леводопой.Поскольку полный ответ на леводопу может быть диагностическим для DRD и из-за его потенциальной пользы, всем детям с дистонией настоятельно рекомендуется испытание карбидопы / леводопы. 140
Лечение ботулиническим токсином — это лечение первого выбора при большинстве типов очаговой дистонии. У детей он используется реже, потому что дистонические формы в основном являются генерализованными, но он также может быть полезен для контроля наиболее инвалидизирующих симптомов сегментарной или генерализованной дистонии. 141 , 142 Ботулинический токсин может рассматриваться как лечение первой линии при первичной краниальной (за исключением оромандибулярной) или шейной дистонии.Ботулинический токсин безопасен и эффективен при повторных курсах лечения в течение многих лет, но чрезмерные кумулятивные дозы могут быть опасными, особенно у детей. После предварительных предположений о том, что интратекальное введение баклофена может улучшить дистонию, последующие наблюдения показали, что эта форма лечения особенно полезна, когда спастичность сосуществует с дистонией, например, при церебральном параличе. 143 — 146
Долгосрочная электростимуляция внутреннего бледного шара (GPi) в настоящее время признана эффективным средством лечения различных типов двигательных нарушений, включая дистонию.Использование DBS при дистонии в настоящее время касается, в частности, первичных генерализованных или сегментарных форм у пациентов, которые не достигают достаточного облегчения при консервативных подходах. Паллидный DBS считается хорошим вариантом, особенно при первичной генерализованной или сегментарной дистонии, после того, как лекарства или ботулотоксин не привели к адекватному улучшению. Было замечено, что пациенты с дистонией DYT1 имеют лучший результат, чем пациенты с отрицательным результатом DYT1 , и что чем раньше будет проведено вмешательство в течение болезни, тем лучше результат. 147 , 148 DBS особенно эффективен у детей, страдающих генерализованной дистонией. Невозможно показать, что возраст пациента имеет прямое прогностическое значение для результата. 148 — 150 Более короткая продолжительность болезни — независимо от ее начала — усиливает эффект стимуляции. Эти данные поддерживают раннее вмешательство. Таким образом, при ранней дистонии у детей не следует откладывать возрастное вмешательство.Предварительная информация о небольшом количестве пациентов с M-D, получавших паллидальный DBS, указывает на то, что как миоклонические признаки, так и дистония могут улучшиться. 151
В обзоре 109 пациентов с наследственно-дегенеративными и вторичными синдромами, получавших имплантаты GPi-DBS, некоторые случаи с благоприятными исходами были зарегистрированы почти во всех группах. 152 Кроме того, у восьми из 13 пациентов с PKAN отмечалось резкое улучшение моторики. 153
Обновленная информация о детских дистониях: этиология, эпидемиология и лечение
Реферат
Дистония — это двигательное расстройство, характеризующееся устойчивыми мышечными сокращениями, вызывающими скручивание, повторяющиеся и шаблонные движения или неправильные позы.Дистония — одно из наиболее часто наблюдаемых двигательных расстройств в клинической практике как у взрослых, так и у детей. Он классифицируется на основе этиологии, возраста появления симптомов и распределения пораженных участков тела.
Этиология
Этиология детской дистонии весьма неоднородна. Существует множество различных генетических синдромов и несколько причин симптоматических синдромов. Дистония может быть вторичной по отношению практически к любому патологическому процессу, поражающему двигательную систему, и особенно базальные ганглии.
Классификация
Этиологическая классификация различает первичную дистонию без идентифицированной экзогенной причины или признаков нейродегенерации и вторичных синдромов.
Лечение
Лечение большинства форм дистонии симптоматическое и включает в себя лекарственные препараты (системные или очаговые методы лечения, такие как ботулинический токсин) и хирургические процедуры. Есть несколько лекарств, в том числе антихолинергические, дофамин-блокирующие и истощающие агенты, баклофен и бензодиазепины.У пациентов с нарушениями синтеза дофамина лечение L-допа может быть очень полезным. Лечение ботулиническим токсином может быть полезным в борьбе с наиболее ограничивающими симптомами сегментарной или фокальной дистонии. Длительная электрическая стимуляция внутреннего бледного шара оказывается особенно успешной у детей, страдающих генерализованной дистонией.
Ключевые слова: двигательные расстройства, детская дистония, первичные дистонии, вторичные дистонии
Введение
Дистония — это двигательное расстройство, характеризующееся устойчивыми мышечными сокращениями, вызывающими скручивание, повторяющиеся и шаблонные движения или аномальные позы.Дистония имеет широкий клинический спектр: от минимальных или доброкачественных самоограничивающихся признаков до тяжелых случаев. 1 — 3 Дистония классифицируется на основе: (i) этиологии, (ii) возраста появления симптомов и (iii) распределения пораженных участков тела. Этиология детской дистонии неоднородна (). 4 Этиологическая классификация различает первичную дистонию без идентифицированной экзогенной причины или признаков нейродегенерации и вторичных синдромов.Первичные заболевания можно разделить на чистые, плюс и пароксизмальные. В первичных чистых формах дистония является единственным признаком заболевания (за исключением тремора), а причина либо генетическая, либо неизвестная. Первичные плюсовые синдромы характеризуются дистонией, связанной с дополнительным двигательным расстройством (например, миоклонусом или паркинсонизмом). Наконец, первичные пароксизмальные синдромы включают состояния, характеризующиеся аномальными движениями, включая дистонию, возникающую в виде коротких эпизодов с нормальным состоянием между ними.Вторичные дистонические формы включают синдромы, при которых дистония является ярким признаком наследственно-дегенеративного состояния или когда дистония вызвана экзогенными факторами (например, перинатальная травма, лекарства, опухоль головного мозга, инфекции).
Таблица 1
Классификация дистоний по этиологии
| Первичная | |
|---|---|
| • Первичная чистая | Дистония является единственным клиническим признаком (кроме тремора), и нет никакой идентифицируемой экзогенной причины или другое наследственное или дегенеративное заболевание |
| • Первичная плюс | Дистония является заметным признаком, но связана с другим двигательным расстройством, например миоклонусом или паркинсонизмом.Нет свидетельств нейродегенерации. |
| • Первичная пароксизмальная | Дистония возникает короткими эпизодами с нормальным состоянием между ними. Эти расстройства классифицируются как идиопатические (часто семейные, хотя встречаются и спорадические случаи) и симптоматические из-за множества причин |
| Вторичные | Дистония является симптомом определенного неврологического состояния, такого как очаговое поражение мозга, воздействие лекарств. или химические вещества |
| Наследственная дегенеративная | Дистония — это особенность, среди других неврологических признаков, наследственно-дегенеративного или метаболического расстройства |
Вторая ось классификации различает начало у детей (<18 лет) и начало у взрослых (> 19 лет) кейсы ().Наконец, третья ось, основанная на соматическом распределении дистонии, различает фокальные, сегментарные, многоочаговые, генерализованные и односторонние (гемидистония) формы ().
Таблица 2
Классификация дистоний по возрасту начала
| Раннее начало (варьируется от 20 до 30 лет) | Обычно начинается с конечности и часто распространяется на другие конечности и туловище |
| Позднее начало | Обычно начинается с шеи (включая гортань), черепных мышц или одной руки.Имеет тенденцию оставаться локализованной с ограниченным прогрессированием в соседние мышцы |
Таблица 3
Классификация дистоний по распределению
| Очаговая | Отдельная область тела |
| Сегментарная | Смежные области тела |
| Многоочаговая | Несмежные области тела |
| Обобщенный | Обе ноги и хотя бы одна другая часть тела |
| Гемидистония | Половина тела |
Дистонические синдромы относятся к наиболее часто наблюдаемым двигательным расстройствам в клинической практике как у взрослых, так и у детей, с распространенностью от 2 до 50 случаев на миллион для дистонии с ранним началом (<26 лет) и от 30 до 7320 случаев на миллион для дистонии с поздним началом (> 26 лет). 5 — 7
В этом обзоре мы сосредоточимся на дистонии, которая поражает детей, произвольно ограничиваясь лицами моложе 18 лет. Мы обсудим первичные дистонии с педиатрическим началом, разделенные на «чистые» дистонии (DYT1, DYT6, DYT13, DYT17), «дистонии плюс», связанные с миоклонусом (DYT11 и DYT15), и «дистонии плюс», связанные с паркинсонизмом (DYT5). , DYT12, DYT16). Наконец, мы включим наиболее частые вторичные дистонии.
Синдромы первичной чистой дистонии
В чистом виде дистония является единственным клиническим признаком (кроме тремора).Детская первичная чистая дистония — редкое заболевание; сюда входят семейные и спорадические случаи. Было картировано семь локусов для первичной дистонии, начинающейся у детей, включая DYT1, DYT4, DYT6, DYT13 и DYT17 (). Среди них на данный момент известны только два причинных гена, DYT1 / TOR1A и DYT6 / THAP1. 8
Таблица 4
Молекулярная классификация непароксизмальных дистоний, начинающихся у детей
| Ген | Локус | Обозначение | Продукт гена | Тип |
|---|---|---|---|---|
| С аутосомно-доминантным наследованием | ||||
| DYT1 | 9q34 | Идиопатическая торсионная дистония (ITD) | Торсин A ( TOR1A ) | Первичный чистый |
| DYT4 | Неизвестно | Идиопатическая торсионная дистония | Неизвестная торсионная дистония Неизвестная торсионная дистония 4949||
| DYT5 | 14q22.1 | Дефицит GTPCH | GTP-циклогидролаза 1 ( GCh2 ) | Первичный плюс |
| DYT6 | 8p11–21 | Подростковое / раннее взрослое начало | N-концевой домен THAP ( THAP1 ) | Первичный чистый |
| DYT11 | 7q21 AD | Миоклоническая дистония | ε-саркогликан ( SGCE ) | Первичный плюс |
| DYT12 | 9q13 AD | Rapid-onset + / K + -ATPase ( ATP1A3 ) | Основной плюс | |
| DYT13 | 1p36.13–36,32 | Дистония с ранним началом с поражением шейного отдела черепа и верхних конечностей | Неизвестно | Первичная чистая |
| DYT15 | 18p11 | Миоклоническая дистония, чувствительная к алкоголю | Неизвестно | |
| DYT16 | 2q31.2 | Дистония-паркинсонизм с молодым началом | Киназа PKR ( PRKRA ) | Первичная плюс |
| DYT17 | 20p11.22-q13.12 | Аутосомно-рецессивный первичный очаговый TD | Неизвестно | Первичный чистый |
| С Х-сцепленным наследованием | ||||
| DYT3 | Xq13.1 | Х-сцепленная дистония / паркинсонизм | Фактор 1, связанный с TATA-связывающим белком ( TAF1 ) | Первичный плюс |
| Xq13 | X-связанный синдром дистонии-глухоты (синдром Мора-Транебьерга) | Импортная транслоказа внутренней мембраны митокондрий Tim8A ( TIMM8A ) | ||
DYT1 (
TOR1A ): Дистония с ранним началомДистония с ранним началом, вызванная мутацией DYT1 , представляет собой наиболее частую и наиболее тяжелую форму первичной дистонии.На его долю приходится 16–53% случаев детской дистонии (POD) среди нееврейского населения и 80–90% пациентов среди еврейского населения ашкенази. 9 — 11
Рассчитанная частота заболевания среди еврейского населения ашкенази составила 1: 3000–1: 9000. Среди нееврейского населения частота примерно в пять раз ниже. 12 , 13
Обычно дистония DYT1 начинается в детстве или подростковом возрасте. Первоначальным симптомом обычно является дистония очагового действия, затрагивающая одну конечность (писчая дистония, ходячая дистония с инверсией или выворотом стопы).Дистония впоследствии распространяется на другие области тела, становится менее специфичной и может также присутствовать в состоянии покоя. Пациенты с дистонией ног в начале болезни обычно имеют более раннее начало (средний возраст 9 лет) и генерализованную дистонию через несколько месяцев или лет. Пациенты с дистонией руки имеют более позднее начало (средний возраст 15 лет), и генерализация дистонии встречается реже. Дистония также может начаться в шее или черепных мышцах. 14
В целом, до 65% пациентов с дистонией DYT1 имеют генерализованную дистонию, при которой чаще всего поражаются конечности.Черепные мышцы поражаются реже, примерно у 11–18% пациентов. Пораженные члены семьи могут иметь начало у взрослых и чаще с поражением шеи. Сообщалось о необычных фенотипах, таких как изолированный блефароспазм и односторонняя миоклоническая дистония. 15
У некоторых пациентов с дистонией DYT 1 может наблюдаться резкое ухудшение дистонии, называемое «status dystonicus», в течение болезни. 16 Продукт гена DYT1 , TorsinA, является членом суперсемейства АТФаз AAA1, связанных с различными клеточными активностями.Он выполняет важные функции, связанные с деградацией белков, переносом мембран, слиянием везикул и перемещением органелл, динамикой цитоскелета и правильным сворачиванием белков. 17
TorsinA экспрессируется почти повсеместно, и его экспрессия в головном мозге ограничена нейронами, где он связан с эндоплазматическим ретикулумом (ER). В клеточных моделях, экспрессирующих патогенную делецию GAG, мутантный TorsinA перераспределяется из просвета ER в ядерную оболочку (NE). 18 Эти клетки также обнаруживают аномальную морфологию и утолщение NE, включая образование включений мутовидной мембраны, которые, по-видимому, происходят из ER и NE. Эти включения связаны с везикулярным транспортером моноаминов VMAT2, открытие, которое может функционально связать TorsinA с дофаминергической системой. 19 Кроме того, было обнаружено, что TorsinA регулирует клеточный перенос переносчика дофамина и других мембраносвязанных белков. Также было показано, что мутант TorsinA вмешивается в процессы цитоскелета, которые могут влиять на развитие нейрональных путей в головном мозге, и что он преждевременно разрушается как протеасомными путями, так и путями макроаутофагии. 20 Недавно было показано, что TorsinA контролирует стабильность белков синаптических везикул и влияет на оборот синаптических везикул в нейронах. 21
Первичные дистонии с ранним началом, не связанные с DYT1
Большая группа пациентов с ПОЛ не связана с мутацией DYT1 . DYT6 дистония была описана в двух семьях германо-меннонитского происхождения. 22 Фенотип определяется как смешанный, потому что он отличается от типичного педиатрического начала: варьирующий возраст в начале от младенчества до взрослого возраста (диапазон 5–38 лет; среднее значение 18.6 лет), выраженное поражение черепных и шейных мышц, генерализация дистонии наблюдается примерно у половины пациентов. 23 Продукт гена DYT6 , Танатос-ассоциированный белок 1 (THAP1), является членом семейства клеточных факторов, разделяющих высококонсервативный ДНК-связывающий домен THAP1, который представляет собой атипичный цинковый палец и может регулировать эндотелиальные клетки. распространение. 24 Предполагаемый механизм заболевания заключается в том, что мутации DYT6 могут нарушать связывание THAP1 с ДНК и вызывать нарушение регуляции транскрипции.Недавно было продемонстрировано, что между THAP1 и промотором TOR1A происходит физическое взаимодействие, которое устраняется патогенными мутациями. 25
Эта связь, вероятно, не просто простое взаимодействие промоторов THAP1-Tor1A, но, вероятно, также включает другие гены. Интересно, что THAP1 также связывается с промоторной областью транскрипта TAF1, который участвует в форме первичной плюс дистонии (Х-сцепленная паркинсонизм с началом Х-хромосомы). 26
Другой редкой причиной раннего начала первичной дистонии является мутация DYT13 , которая была обнаружена в большой итальянской семье. 27 Преобладающим фенотипом является детская дистония верхней части тела с частым поражением черепно-шейной области и относительно доброкачественным течением.
Первичная дистония DYT17 описана как пример аутосомно-рецессивного POD. 28 , 29 Локус для DYT17 был назначен на хромосому 20.
Первичные плюсовые синдромы
Дистония является заметным признаком дистонии плюс синдромов, но она связана с другими двигательными расстройствами (например, myoclonus или паркинсонизм) без признаков нейродегенерации.Выделяют три клинически определяемых состояния: допа-зависимая дистония ( DRD; DYT5 ), миоклоническая дистония (MD; DYT11 ) и быстро развивающаяся дистония-паркинсонизм (RDP; DYT12 ), которые характеризуются выраженным фенотипическая гетерогенность.
DRD или болезнь Сегавы
DRD или болезнь Сегавы в основном возникает у детей из-за дефектов синтеза дофамина. Обычный возраст начала заболевания колеблется от 4 до 6 лет. Соотношение самок и самцов — 2.5: 1. 30 Сообщалось о распространенности от 0,5 до 1 на миллион, но, вероятно, она недооценена, поскольку пенетрантность низкая, а атипичные случаи часты. 31 Начало часто коварное, с утомляемостью, неуклюжей походкой и дистоническими позами, часто ограниченными одной стопой. 32 , 33
У детей старше 10 лет первыми проявлениями могут быть дистония верхних конечностей или постуральный тремор. 34 Обычно присутствуют легкие признаки паркинсонизма, такие как ригидность конечностей и туловища и гипомимия.Очень характерно прогрессирующее усиление дистонии в течение дня и заметное уменьшение, а иногда и полное исчезновение после сна. Расстройство прогрессирует и может привести к тяжелой инвалидности. Поражение других частей тела может произойти быстро или занять до десяти лет. Когнитивные функции сохранены.
Дистония и / или паркинсонизм обычно показывают быстрый, выраженный и устойчивый ответ на низкие дозы (3-5 мг / кг / день) L-допа в сочетании с ингибитором периферического декарбоксилирования, помогающим установить клинический диагноз.Ответ не зависит от задержки в начале лечения. Дефицит аутосомно-доминантной гуанозинтрифосфатциклогидролазы (GTPCH) ( DYT5 ) является наиболее частым ферментативным дефектом, приводящим к болезни Сегавы. Почти 75% случаев индекса DRD большой европейской серии показали этот дефект. 35
Сообщалось об аутосомно-рецессивных формах GTPCH. 36 Аутосомно-рецессивные формы DRD (ArDRD) также возникают в результате гомозиготных или сложных гетерозиготных мутаций тирозингидроксилазы (TH) и сепиаптеринредуктазы (SPR). 37 При ArDRD фенотип обычно более тяжелый, включая когнитивные нарушения и задержку развития, и его также можно назвать инфантильным энцефалопатическим фенотипом (IEP). 3 Ответ на лечение L-допа при IEP отличается от болезни Сегавы. Только в редких случаях низкие дозы L-допы могут привести к быстрому улучшению симптомов. У большинства этих пациентов низкие дозы L-допы могут вызывать выраженные хореические и / или дистонические движения. У большинства этих пациентов необходимо очень медленное титрование дозы, и часто невозможно достичь эффективных терапевтических уровней L-допа.Даже низкие дозы могут вызывать заметные хореические и / или дистонические движения. Прогноз ИЭП отличается от прогноза болезни Сегавы даже в тех случаях, когда частично отвечает дофаминергическая терапия; Часто сообщается о неуклюжести и легкой умственной отсталости.
Недавно у некоторых пациентов с ИЭП, вызванным мутациями в гене, кодирующем переносчик допамина ( SLC6A3 ), было зарегистрировано новое состояние, называемое синдромом дефицита переносчика дофамина (DTDS). Исследования нейротрансмиттеров спинномозговой жидкости показали повышенное соотношение HVA / HIAA.Сканирование переносчика дофамина с помощью однофотонной эмиссионной компьютерной томографии (DaTSCAN SPECT) показывает полную потерю активности переносчика дофамина в базальных ядрах. 38
Миоклоническая дистония (M-D) (
DYT11 )Заболевание начинается в детском или подростковом возрасте. Симптомом обычно является миоклонус, поражающий верхнюю часть тела (шею, туловище, конечность) с преобладанием проксимальных мышц. Также могут быть затронуты черепные мышцы, в том числе мышцы гортани. 39 — 43 Дистония связана с миоклонусом более чем у половины пациентов и обычно вызывает незначительную инвалидность или не вызывает ее вовсе. Дистония обычно поражает шею или руки (кривошея, судорога писателя). 40 , 41 , 44 — 46 Заметное улучшение двигательных симптомов (в основном миоклонуса) после приема алкоголя и наличия психических симптомов, таких как обсессивно-компульсивные расстройства, злоупотребление алкоголем часто сообщается о депрессии и тревоге. 44 , 47 — 49 Течение болезни, как правило, доброкачественное, но исходы у пациентов, даже в пределах одной семьи, сильно различаются. 41
M-D вызывается гетерозиготными мутациями гена эпсилон-саркогликана ( SGCE ). Выявлено более 50 различных гетерозиготных мутаций SGCE . 50 Считается, что эти мутации вызывают потерю функции и не позволяют провести корреляцию генотип-фенотип.Однако заметные различия в степени тяжести заболевания предполагают влияние факторов окружающей среды или генетических модификаторов. Геномной особенностью SGCE является материнский геномный импринтинг. 51
Дистония-паркинсонизм с быстрым началом (RDP,
DYT12 )Заболевание характеризуется внезапной дистонией и паркинсонизмом. 52 , 53 В своей генетической форме тип наследования аутосомно-доминантный с пониженной пенетрантностью, и сообщалось о мутациях de novo. 54 , 55 Одной из моногенетических причин RDP является мутация в гене альфа-субъединицы Na + / K + -АТФазы 3 (a3; ATP1A3 ). Хотя зарегистрировано всего несколько семей и единичные случаи с этим заболеванием, клинические особенности делают это заболевание особенно интересным. Расстройство обычно начинается в возрасте от 15 до 45 лет. Отличительными признаками являются резкое появление двигательных симптомов в течение от нескольких минут до 30 дней с вовлечением рострокаудальной области и выраженными бульбарными находками.Начало часто следует за физическим или эмоциональным стрессом, приводящим к стойкой неврологической инвалидности. 54 Большинство пациентов остаются стабильными или демонстрируют небольшое улучшение через годы после резкого появления симптомов. Заболевание передается по аутосомно-доминантному признаку с переменной выраженностью и пониженной пенетрантностью. Мутации в гене ATP1A3 (хромосома 19q13), связанном с NA + / K + ATPase alpha3, являются причиной нарушения. 54 , 55 RDP — первое известное дистоническое состояние, связанное с аномалией мембранного ионного канала.Все исследования остаются отрицательными, за исключением низкого уровня HVA в спинномозговой жидкости, что свидетельствует о пресинаптическом дефекте нигростриатных путей. 56
Недавно был идентифицирован новый ген PRKRA (протеинкиназа, интерферон-индуцибельный двухцепочечный РНК-зависимый активатор) для молодой дистонии-паркинсонизма ( DYT16 ). 57
Другие первичные генерализованные дистонии
Х-связанная дистония глухоты
Сообщалось о нескольких семьях с синдромом Х-связанной дистонии-глухоты (также называемым синдромом Мора – Транебьерга). 58 , 59 Заболевание характеризуется началом сенсоневральной глухоты обычно в возрасте до 2 лет. Тяжелая прогрессирующая генерализованная дистония начинается примерно в 7-летнем возрасте. Сообщалось о случаях развития дистонии уже в возрасте 30 лет, что позволяет предположить, что клинические признаки имеют более широкий спектр, чем считалось ранее. 60 , 61 Большинство испытуемых приковывались к инвалидной коляске в возрасте от 9 до 22 лет.Умственная отсталость, нарушение зрения из-за корковой слепоты или связанное с нейропатией зрительного нерва, 62 вовлечение кортикоспинального тракта и психиатрические проявления (раздражительность, беспокойство, паранойя) могут быть ассоциированными признаками. Ген-кандидат для этого расстройства, названный DDP (пептид глухоты / дистонии) или TIMM8A (вероятно, кодирующий импортируемую в митохондрии субъединицу транслоказы внутренней мембраны Tim8 A, цинк-связывающий белок), был идентифицирован на хромосоме Xq22. 63 Белок (белок DDP) расположен в межмембранном пространстве митохондрий, поэтому мутированный белок DDP может нарушить функцию митохондрий. 64 Сообщалось о нескольких мутациях, включая интронные. 65 По крайней мере, в одной из описанных мутаций самки-носители также страдают дистонией, но, в отличие от мужчин, она описывается как очаговая дистония. 66
ГАМКергические вещества, такие как клоназепам и гаммагидроксимасляная кислота, считаются полезными. 60
Преходящая идиопатическая дистония младенчества
Преходящая дистония младенчества обычно проявляется в возрасте до 5 месяцев. 67 , 68 У пораженных младенцев аномальные позы обычно ограничиваются одной верхней или нижней конечностью. Иногда в позы могут вовлекаться туловище, руки или одна сторона тела. 69 В положении лежа младенец часто поддерживает форсированную пронацию предплечья, используя тыльную сторону кисти в качестве опоры. В нижней конечности ступни могут находиться в эквиноварусии. Позы могут быть прерывистыми. 70 В остальном неврологическое обследование и развитие в норме.Двигательное расстройство самопроизвольно исчезает к первому дню рождения. Семейные случаи известны. 67 , 68 Легкие формы могут быть частыми, и только в наиболее выраженных случаях может потребоваться медицинская помощь. Ключом к диагнозу является наблюдение, что дистоническая поза исчезает, когда младенец выполняет целенаправленные движения пораженной конечностью. Этиология пока неизвестна.
При типичных клинических признаках дополнительные исследования (нейровизуализация, лабораторные исследования) не требуются.
Первичные пароксизмальные синдромы
Пароксизмальные дискинезии относятся к приступам аномальных движений и поз (дистонических, хореоатетоидных или их комбинации) с возвращением к нормальному состоянию между эпизодами. 71
Можно выделить три основные категории: пароксизмальная кинезигенная дискинезия (PKD / DYT10), пароксизмальная некинезигенная дискинезия (PNKD1; DYT8) и пароксизмальная дискинезия, вызванная физической нагрузкой (PED; DYT18). 72 В ДОК продолжительность атак варьируется от секунд до часов, а частота эпизодов варьируется от одного в год до сотен в день.PKD характеризуется дистонией или хореодистонией, вызванной внезапной сменой положения, обычно переходящего из положения сидя в положение стоя. PNKD характеризуется спонтанными приступами в покое, которые имеют тенденцию к более дистоническому характеру, хотя могут возникать хорея, атетоз и баллизм. Эпизоды могут длиться от секунд до нескольких часов и иметь длинные интервалы без приступов. Начало в детстве с тенденцией к уменьшению приступов с возрастом. 73 Симптомы могут быть спровоцированы алкоголем или кофеином и, в меньшей степени, никотином, возбуждением, усталостью, голодом и эмоциональным стрессом.Пароксизмальный хореоатетоз с клиническими характеристиками, подобными таковым для PNKD с ассоциированной спастичностью, был описан под термином аутосомно-доминантный пароксизмальный хореоатетоз / синдром спастичности (CSE), и была продемонстрирована связь с хромосомой 1p. 74 В PED приступы возникают после 10 или 15 минут непрерывных упражнений. Приступы обычно дистонические и чаще всего появляются в нижних конечностях после продолжительной ходьбы или бега. Приступы дистонии обычно прекращаются через 10–15 минут после прекращения упражнений. 75
Пациенты с мутациями гена SLC2A1 (ген, кодирующий транспортер Glut 1) могут иметь PED, который может быть связан с эпилепсией и / или умственной отсталостью. 76 — 78
Список локусов сцепленных генов, вызывающих фенотипы пароксизмальной дискинезии, быстро растет, хотя гены большинства этих состояний все еще остаются неидентифицированными. Пароксизмальные дискинезии наследуются по аутосомно-доминантному признаку. 79 PNKD вызывается мутациями в гене PNKD1 , кодирующем регулятор 1 миофибриллогенеза.При PKD локус заболевания был приписан перицентромерной области хромосомы 16 (16p11.2-q12.1). 80 Первый ген, вызывающий PKD, был недавно идентифицирован: трансмембранный белок пролинерих 2. 81 , 82 Ген PED расположен в коротком плече хромосомы 1 (1p31. 3 – p35). Ген SLC2A1 кодирует транспортер глюкозы 1 (Glut1). 77 Поскольку Glut1 доставляет глюкозу через гематоэнцефалический барьер, дискинезии могут быть результатом дефицита энергии в базальных ганглиях при физической нагрузке.
Вторичные дистонии
Дистония часто ассоциируется с другими неврологическими симптомами в длинном списке заболеваний, и затем ее называют вторичной дистонией.
Дистония, вызванная структурным дефектом
Дистонический церебральный паралич и дистония с отсроченным началом не будут обсуждаться в этом обзоре. Гемидистония часто возникает из-за структурного поражения. Наиболее частой причиной гемидистонии является острая окклюзия сосудов любой этиологии, особенно поражающая полосатое тело и / или хвостатое тело. 83 Гемидистония чаще встречается у детей, чем у взрослых. 84 Гемидистония преобладает в верхних конечностях и очень часто сопровождается признаками пирамидного тракта. Первым неврологическим проявлением обычно является острый гемипарез. Дистония может появиться во время выздоровления гемипареза, но часто она становится очевидной только через несколько месяцев или даже лет. 84
Часто оказывается плохой ответ на лечение. Наилучшие результаты дают бензодиазепины и антихолинергические препараты.Может быть полезен ботулинический токсин. Хирургия, в основном глубокая стимуляция головного мозга (DBS) в определенных областях внутреннего бледного шара, кажется сейчас лечением выбора. 85
Наследственные дегенеративные синдромы
Наследственные дегенеративные синдромы составляют большую и разнородную группу заболеваний, при которых дистония может возникать с разной степенью тяжести и связана с другими аномалиями, включая умственную отсталость, судороги, атаксию, невропатию, атрофию зрительного нерва и взор. парез.Многие из этих расстройств являются аутосомно-рецессивными из-за наследственных дегенеративных или метаболических заболеваний и возникают в основном в детстве.
Глутаровая ацидурия и другие органические ацидемии с дистонией
Глутаровая ацидурия I типа (GAI), также называемая дефицитом глутарил-КоА-дегидрогеназы, представляет собой аутосомно-рецессивное заболевание, вызываемое мутациями в гене глутарил-КоА-дегидрогеназы ( GCDH), расположенном на хромосоме GCDH. 19п13.2. 86 , 87 GCDH — митохондриальный фермент, который играет ключевую роль в катаболизме лизина, гидроксилизина и триптофана.Сильное снижение или полное отсутствие активности GCDH вызывает накопление 3-гидроксиглутаровой кислоты и глутаровой кислоты в плазме, моче и спинномозговой жидкости, что может вызвать гибель нейронов из-за эксайтотоксичности, а также дисфункции митохондрий. 88
Частота ГАИ оценивается в 1 на 100 000 новорожденных. 89 Клинические проявления обычно появляются в возрасте от 5 до 14 месяцев, но легкие симптомы, такие как небольшая задержка моторики и гипотония, могут наблюдаться в более раннем возрасте.Макроцефалия при рождении или развившаяся позже в младенчестве присутствует примерно в 70% случаев. 90 , 91 В большинстве случаев заболевание начинается внезапно, с очаговыми припадками или генерализованными судорогами, рвотой и заторможенностью или летаргией, обычно после острого инфекционного заболевания. Позже появляются психомоторный регресс и дистонические или хореоатетотические движения. У некоторых пациентов начало незаметно с медленно развивающейся задержкой психомоторного развития, гипотонией и дистоническими позами до 16 лет. 92 — 95
Характерным неврологическим последствием является генерализованная дистония. Также может быть очевидна спастичность. Часто встречаются сложные речевые изменения с сочетанием признаков гиперкинетической дизартрии, анартрии и апраксии. Понимание языка намного лучше, чем выражение, что говорит о том, что познание относительно сохранено.
Течение расстройства непостоянно. ГАИ — излечимое состояние. В идеале лечение следует начинать до появления клинических симптомов.В таких случаях диета в сочетании с пероральным приемом L-карнитина и агрессивным лечением во время острых эпизодов интеркуррентного заболевания может предотвратить развитие клинических признаков или уменьшить их тяжесть. Однако в большинстве случаев болезнь продолжает прогрессировать поэтапно с чередой острых энцефалопатических кризов, вызванных инфекционными заболеваниями, вакцинацией и хирургическим вмешательством в младенчестве или детстве. Часто с возрастом дистония имеет тенденцию эволюционировать от мобильной к стационарной и ассоциироваться с паркинсонизмом. 96 В других случаях он остается неподвижным с тяжелыми двигательными и языковыми последствиями. 97 Около половины пациентов умирают в возрасте до 4 лет в результате интеркуррентных заболеваний.
Его патогенез не ясен. Концентрации глутаровой и 3-гидроксиглутаровой кислоты увеличиваются не только в головном мозге, но и во всех тканях. 3-гидроксиглутаровая и глутаровая кислоты имеют структурное сходство с глутаматом основной возбуждающей аминокислоты, и считается, что повышенные уровни этих кислот играют важную роль в патофизиологии заболевания.3-гидроксиглутаровая кислота вызывает эксайтотоксическое повреждение клеток. Кроме того, глутаровая и 3-гидроксиглутаровая кислоты косвенно модулируют глутаматергическую и ГАМКергическую нейротрансмиссию (что приводит к низким уровням ГАМК), что приводит к дисбалансу возбуждающей и тормозящей нейротрансмиссии. 88
Спектр Леша – Найхана
Спектр Леша – Найхана представляет собой Х-сцепленное нарушение метаболизма пуринов, возникающее в результате дефицита фермента гипоксантин-гуанинфосфорибозилтрансферазы (HGPRT), расположенного на Xq26. 98 , 99 Возможна пренатальная диагностика.
Существует непрерывный спектр неврологических проявлений в зависимости от степени ферментативной недостаточности. Наиболее тяжелые формы известны как болезнь Леша – Нихана (ЛБД). У пациентов с частичным дефицитом HPRT симптомы различаются по интенсивности. В наименее тяжелой форме, называемой синдромом Келли-Сигмиллера, у пациентов наблюдается подагра без неврологических нарушений. 100 Термин «варианты Lesch-Nyhan» был введен для обозначения пациентов с подагрой, связанной с HPRT, и некоторой степенью неврологического поражения, но без полной симптоматики LND.
LND обычно начинается в возрасте от 6 до 18 месяцев с задержкой психомоторного развития, гипотонией или спастичностью. Ранним признаком может быть появление в подгузниках кристаллического вещества оранжевого цвета. Аномальные движения начинаются с тонких атетоидных движений кистей и стоп. В полностью развитом состоянии движения преимущественно дистонические, но могут включать хорею и тремор. На моторику глаз влияют саккады, которым предшествуют движения головы. 101 Примечательной особенностью является агрессивное поведение (85% случаев), направленное как на самого пациента, так и на окружающих.Автоматизация, особенно поражающая губы и пальцы, обычно начинается с прорезывания зубов. Дизартрия также является частым симптомом. Сообщалось об эпилепсии, атрофии зрительного нерва и рецидивирующей коме. 102 , 103 У больных детей также наблюдаются экстраневрологические симптомы, такие как гематурия, кристаллурия и признаки гиперурикемии, такие как почечные камни, подагрический артрит и тофусы. Другие особенности включают рвоту, макроцитарную анемию, замедленный рост и задержку костного возраста.Сейчас часты случаи, когда они выживают после третьего десятилетия жизни.
В вариантах Леша – Нихана степень дистонии менее тяжелая и проявляется в виде дистонической походки, затруднений речи и спастичности, но с нормальными когнитивными функциями и поведением (без членовредительства).
Патофизиология неврологических и поведенческих нарушений остается неясной. Предполагается, что неврологические симптомы не являются результатом избыточного производства мочевой кислоты. Патологоанатомические исследования не выявили каких-либо структурных изменений, но выявили потерю дофамина в базальных ганглиях на 60–90%. 104 Биохимические исследования спинномозговой жидкости и ПЭТ также показали снижение уровня метаболитов дофамина, тогда как серотонин и 5-гидроксииндолуксусную кислоту были увеличены. 105
Гомоцистинурия
Гомоцистинурия вызывается различными ферментативными дефектами. Чаще всего встречается дефицит цистатионин-β-синтазы. Сообщалось о нескольких случаях гомоцистинурии с медленно прогрессирующей дистонией как поздним симптомом. 106 , 107 В этих случаях возраст начала дистонии колеблется от 4 до 21 года.
Причина двигательного расстройства не выяснена. Вероятно сосудистое заболевание. При гомоцистинурии сообщалось об артериальных и венозных тромбозах коры головного мозга и базальных ганглиев, но не было описано нейровизуализационных доказательств поражения базальных ганглиев в случаях дистонии. Кроме того, при патологоанатомическом исследовании сосудов базальных ганглиев патологии не выявлено. 108 , 109
Болезнь Хартнупа
Болезнь Хартнупа является наследственным рецессивным дефектом транспорта аминокислот из-за мутаций в гене SLC6A19 . 110 Светочувствительный дерматит обычно является первым симптомом, возникающим в позднем младенческом или юношеском возрасте. Рецидивирующие эпизоды атаксии — частый неврологический симптом. Сообщалось о дистонических особенностях и перемежающейся дистонии. 111 Возможно самопроизвольное улучшение. Терапия никотинамидом может улучшить неврологические симптомы.
Болезнь Ниманна – Пика, тип C (NPC)
Болезнь Ниманна – Пика, тип C — аутосомно-рецессивное нарушение накопления липидов, которое характеризуется нарушенным внутриклеточным гомеостазом холестерина и дефектным перемещением холестерина через позднюю эндосомальную / лизосомную систему. 112 Возникающее в результате накопление неэтерифицированного холестерина, преимущественно в клетках селезенки, печени и мозга, приводит к ряду клинических проявлений. 113 К ним относятся гепатоспленомегалия, психические расстройства и неврологические проблемы. Мутировавший ген ( NPC1 ) на хромосоме 18q11–12 отвечает за более чем 90% случаев. 109 Несколько случаев связаны с мутациями в гене HE1 / NPC2 . 114
Заболевание проявляется вертикальным надъядерным параличом взора, особенно при взгляде вниз.Клинические проявления в начале обычно включают снижение интеллекта, дизартрию и нарушение походки. Двигательное расстройство может преобладать и состоит в основном из дистонии. Затем появляются мозжечковые и пирамидные признаки. Эпилепсия встречается примерно у трети пациентов. Органомегалия встречается часто (но не постоянно). 115 Магнитно-резонансная томография (МРТ) головного мозга показывает диапазон от нормальной до легкой задней перивентрикулярной гиперинтенсивности белого вещества.
Анализ костного мозга показывает пенистые клетки или гистиоциты цвета морской волны.NPC можно диагностировать с помощью культуры фибробластов кожи, демонстрируя недостаточную этерификацию холестерина, производного от липопротеинов низкой плотности, или окрашивая свободный внутриклеточный холестерин на филиппин.
Нет специального лечения для NPC. Миглустат, иминосахар, ингибирующий глюкозилцерамидсинтазу, может показать клиническую пользу. 116
Другие неврологические расстройства с дистонией
Инфантильный двусторонний стриатальный некроз, детский таламический некроз
Friede (1975) предложил термин «детский двусторонний стриарный некроз» (IBSN) для обозначения невропатологического синдрома с двусторонней симметричной губкой путамина и хвостатые ядра, а иногда и таламуса. 117 Число случаев увеличилось с переходом от невропатологического к нейровизуализационному синдрому. Поражения теперь можно распознать у живых пациентов с помощью компьютерной томографии (КТ), которая показывает гиподенсированные изображения в пораженных областях 118 и, что лучше, с помощью МРТ, которая показывает повышенный сигнал Т2 в тех же зонах. 119
Прогресс в знаниях о митохондриальных нарушениях и органических ацидуриях показывает, что симметричные стриарные поражения являются обычным явлением при этих заболеваниях, и предполагает, что многие из случаев, зарегистрированных как IBSN, могли быть вызваны метаболическими нарушениями, декомпенсированными острой инфекцией, например, глутаровая ацидурия. 120 Следовательно, те случаи, в которых не было выявлено никакой органической ацидурии и не был зарегистрирован лактоацидоз, вероятно, являются недоказанными митохондриальными нарушениями.
Типичные случаи острого СРКН характеризуются острым началом ригидности, дистонией, хореей, баллизмом и стереотипной реакцией на раздражители, приятные или болезненные, с монотонным плачем, гримасой и чрезмерным растяжением шеи. 121 Эпилепсия встречается редко. У некоторых пациентов симптомы развиваются вскоре после различных инфекций, включая инфекции дыхательных путей и эпидемический паротит, вызванный Mycoplasma pneumoniae. 122 На снимках часто наблюдается обширный отек белого вещества в дополнение к симметричной гиподности полосатого тела. Поражения при МРТ могут исчезнуть или сохраниться со временем. 123 Некоторые пациенты могут умереть вскоре после острой фазы, но обычно наступает постепенное улучшение, в некоторых случаях даже через несколько месяцев. Могут возникнуть такие последствия, как дистония или гемипарез. 118 , 123
Хотя некоторые семейные случаи зарегистрированы, большинство случаев IBSN носят спорадический характер. 124 Патогенез остается неизвестным, но возможен параинфекционный механизм. Сообщалось о случаях острого IBSN, представленного вскоре после стрептоккального фарингита, с высокими титрами стрептококка и антителами, реактивными против компонентов базальных ганглиев, что предполагает аутоиммунную этиологию в этих случаях. 125
Инфантильный таламический некроз
Случаи острого некроза обоих таламусов («острая некротическая энцефалопатия у детей») после острой вирусной инфекции первоначально были зарегистрированы из Японии, где это состояние, по-видимому, преобладает. 126 Сообщалось также о кавказцах. 127 Половина случаев остро началась в возрасте от 6 до 18 месяцев. 126 Гипертермия, кома, судороги и децеребрационные или декортикальные позы обычны в острой стадии. Течение часто бывает тяжелым, приводя к смерти в четверти случаев или к серьезным остаточным нарушениям с квадриплегией, микроцефалией, дистонией и умственной отсталостью, но существует «легкая» форма с избирательным обратимым поражением таламуса и полным выздоровлением. 128 В начале курса двусторонние гиперэхогенные таламические поражения могут наблюдаться с помощью ультразвука. КТ или МРТ покажут мультифокальные симметричные поражения в таламусе, а также обычно выявляют поражения в мосту, среднем мозге, мозжечке и белом веществе. Может присутствовать гиперпротеиноррахия во время острого заболевания. 129 Обычно присутствуют повышенные сывороточные CPK, GOT, GPT и лактатдегидрогеназа, но они редко имеют гипераммониемию или гипогликемию, что важно для дифференциации диагноза синдрома Рейе. 126 Причинные или ассоциированные вирусные инфекции, обычно грипп A, были продемонстрированы в нескольких случаях. 129 , 130 Прогноз лучше у детей старшего возраста и пациентов с низкими значениями GOT, без повреждений ствола головного мозга и нормализации нейровизуализации при последующем наблюдении. 128 Сообщалось об эффективном лечении стероидами. 131 Эти случаи показывают некоторое сходство с таковыми из ISBN, поэтому был предложен термин «детский таламический двусторонний некроз». 127
Биотин-зависимая болезнь базальных ганглиев
Биотин-зависимая болезнь базальных ганглиев вызывается дефицитом переносчика тиамина-2 (hTHTR2) из-за мутаций в гене SLC19A3 . 132 Биотин-зависимая болезнь базальных ганглиев (BBGD) была впервые описана в 1998 году у 10 арабских пациентов, за которой последовали новые генетически подтвержденные случаи, состоящие из подострых энцефалопатических эпизодов, характеризующихся спутанностью сознания, рвотой с последующими судорогами, потерей речи или дизартрией, параличом взгляда и т. Д. дисфагия, дистония и общая скованность, которые в конечном итоге привели к коме и смерти. 133 , 134 Введение высоких доз биотина приводит к частичному или полному улучшению в течение нескольких дней. При отсутствии лечения заболевание может привести к прогрессирующей энцефалопатии. Характерная МРТ головного мозга в основном показывает двусторонний некроз хвостатых ядер и скорлупы и, если они есть, незначительные изменения в таламусе. Сообщалось также о пользе тиамина.
Чередующаяся гемиплегия в детском возрасте
Чередующаяся гемиплегия в детском возрасте (AHC) — это заболевание, проявляющееся в возрасте до 18 месяцев эпизодами гемиплегии, поражающими обе стороны и исчезающими (иногда всего на несколько минут) во время сна.Продолжительность эпизодов может составлять от нескольких минут до нескольких дней. Характерными признаками являются эпизоды смещения или двустороннего паралича, связанного со косоглазием или односторонним нистагмом. Часто наблюдаются задержка развития и эпилепсия. Причина неизвестна, лабораторных или нейровизуализационных маркеров нет. Заболевание считалось формой мигрени, хотя эта гипотеза оспаривается.
При AHC тонические и дистонические эпизоды, которые часто носят односторонний характер, присутствуют в 90% случаев и часто являются первым проявлением. 130 Двигательное расстройство, состоящее из дистонии, миоклонии или хореи, первоначально появляется только во время эпизодов, но имеет тенденцию становиться постоянным. Генерализованная, но асимметричная дистония или хореоатетоз почти постоянны на поздних стадиях. 135 , 136 Флунаризин частично эффективен примерно у половины пациентов. 137
Варианты лечения у детей
Лечение большинства форм дистонии симптоматическое, и его польза может быть неполной или сопровождаться невыносимыми побочными эффектами.Эти варианты включают лекарства (системные или очаговые методы лечения, такие как ботулотоксин) и хирургические вмешательства. Существует несколько методов лечения, включая антихолинергические препараты, дофаминовые блокаторы и истощающие препараты, баклофен и бензодиазепины, которые были рекомендованы на основе небольших неконтролируемых исследований. 138
Среди медицинских вариантов обычно первым выбирают высокие дозы холинолитиков. Однако абсолютная и сравнительная эффективность и переносимость антихолинергических средств при дистонии у детей плохо документирована.Таким образом, нельзя давать никаких рекомендаций по назначению лекарств (точка хорошей практики). 139 Более того, дети обычно переносят более высокие дозы, чем взрослые. Контролируемых исследований эффектов этого типа лечения не проводилось. Отсутствуют доказательства положительного эффекта тетрабеназина, чтобы дать рекомендации по этому типу лечения (точка надлежащей практики). Карбидопа / леводопа — это основа лечения в РРБ. Однако другие формы дистонии могут частично поддаваться лечению леводопой.Поскольку полный ответ на леводопу может быть диагностическим для DRD и из-за его потенциальной пользы, всем детям с дистонией настоятельно рекомендуется испытание карбидопы / леводопы. 140
Лечение ботулиническим токсином — это лечение первого выбора при большинстве типов очаговой дистонии. У детей он используется реже, потому что дистонические формы в основном являются генерализованными, но он также может быть полезен для контроля наиболее инвалидизирующих симптомов сегментарной или генерализованной дистонии. 141 , 142 Ботулинический токсин может рассматриваться как лечение первой линии при первичной краниальной (за исключением оромандибулярной) или шейной дистонии.Ботулинический токсин безопасен и эффективен при повторных курсах лечения в течение многих лет, но чрезмерные кумулятивные дозы могут быть опасными, особенно у детей. После предварительных предположений о том, что интратекальное введение баклофена может улучшить дистонию, последующие наблюдения показали, что эта форма лечения особенно полезна, когда спастичность сосуществует с дистонией, например, при церебральном параличе. 143 — 146
Долгосрочная электростимуляция внутреннего бледного шара (GPi) в настоящее время признана эффективным средством лечения различных типов двигательных нарушений, включая дистонию.Использование DBS при дистонии в настоящее время касается, в частности, первичных генерализованных или сегментарных форм у пациентов, которые не достигают достаточного облегчения при консервативных подходах. Паллидный DBS считается хорошим вариантом, особенно при первичной генерализованной или сегментарной дистонии, после того, как лекарства или ботулотоксин не привели к адекватному улучшению. Было замечено, что пациенты с дистонией DYT1 имеют лучший результат, чем пациенты с отрицательным результатом DYT1 , и что чем раньше будет проведено вмешательство в течение болезни, тем лучше результат. 147 , 148 DBS особенно эффективен у детей, страдающих генерализованной дистонией. Невозможно показать, что возраст пациента имеет прямое прогностическое значение для результата. 148 — 150 Более короткая продолжительность болезни — независимо от ее начала — усиливает эффект стимуляции. Эти данные поддерживают раннее вмешательство. Таким образом, при ранней дистонии у детей не следует откладывать возрастное вмешательство.Предварительная информация о небольшом количестве пациентов с M-D, получавших паллидальный DBS, указывает на то, что как миоклонические признаки, так и дистония могут улучшиться. 151
В обзоре 109 пациентов с наследственно-дегенеративными и вторичными синдромами, получавших имплантаты GPi-DBS, некоторые случаи с благоприятными исходами были зарегистрированы почти во всех группах. 152 Кроме того, у восьми из 13 пациентов с PKAN отмечалось резкое улучшение моторики. 153
Симптомы, диагностика и лечение> Информационные бюллетени> Йельская медицина
Если вы не занимаетесь деятельностью, требующей осознанного движения, например физиотерапией или изучением нового танцевального движения, вы, вероятно, не тратите много времени на размышления о как контролировать свои мышцы.Это потому, что ваш мозг и нервная система слаженно работают вместе, но, хотя это необычно, иногда возникают проблемы. Дистония — это неврологическое двигательное расстройство, которое приводит к нежелательным мышечным сокращениям или спазмам. Непроизвольные скручивания, повторяющиеся движения или неправильные позы, связанные с дистонией, могут повлиять на любого человека в любом возрасте. Движения могут быть медленными или быстрыми, от легких до изнурительных и происходить предсказуемо или случайно. По данным Национальной организации по редким заболеваниям, около 300000 человек в Северной Америке страдают дистонией.
Дистония — многогранное комплексное заболевание. Различные подтипы поражают участки тела, и их симптомы могут значительно отличаться от человека к человеку. На ранних стадиях и в более легких формах дистония может вызывать раздражение. Например, дистония, поражающая только голосовые связки, может означать, что человеку нужно прилагать дополнительные усилия, чтобы говорить. Но другие формы дистонии могут мешать человеку ходить или есть и являются достаточно серьезными, чтобы потребовать хирургического вмешательства.
Если добавить к этому условию еще больше сложности, исследователи не уверены в его причине.Дистония может развиваться по-разному: от генетических мутаций до побочного действия лекарств. Это может быть симптом другого заболевания, например, болезни Хантингтона или Паркинсона. Во многих случаях дистония возникает по неизвестным причинам.
Хотя заболевание неизлечимо, с некоторыми его формами можно хорошо справиться с помощью индивидуальных планов лечения, которые могут включать в себя лекарства, инъекции ботулинического токсина или операцию по глубокой стимуляции мозга (DBS).
В Yale Medicine команда опытных неврологов и нейрохирургов работает вместе, чтобы найти решения для каждого пациента.Ваш врач проведет подробное собеседование и осмотр, чтобы понять ваши симптомы и узнать, как лучше всего вам помочь. «Одна из целей будет заключаться в том, чтобы определить первопричину вашей дистонии и любые методы лечения», — говорит невролог Йельской медицины Кристин Ким, доктор медицины. «Не менее важной целью будет разработка плана лечения ваших симптомов, который будет тщательно адаптирован к вашим индивидуальным потребностям врачом с большим опытом лечения дистонии. Это может включать в себя прием пероральных препаратов, инъекции ботулинического токсина или рассмотрение операции DBS в соответствующих случаях.Лечение часто включает комбинацию подходов ».
Дистония — обзор | Темы ScienceDirect
Локализация и патофизиология
Дистония связана с рядом заболеваний, но часто нет дискретных, идентифицируемых патологических аномалий в головном мозге. При генетических формах дистонии описаны нарушения на многих уровнях клеточной функции. 8 Однако базальные ганглии играют центральную роль во многих формах дистонии.Когда после инсульта наблюдается дистония, наиболее частыми участками поражения являются скорлупа (наиболее часто), бледный шар или таламус. 9, 10 Кроме того, метаболические нарушения, характерной особенностью которых является дистония, часто связаны с поражением базальных ганглиев. Поражение структур базальных ганглиев у приматов может вызывать аномальные позы, характерные для дистонии человека. 11 Связывание рецептора допамина D 2 снижено в скорлупе пациентов с первичной очаговой дистонией и в модели транзиторной дистонии на животных. 12, 13 Эти наблюдения и другие данные предполагают, что дистония возникает из-за аномальной функции базальных ганглиев, что, в свою очередь, вызывает изменение регуляции кортикальных, спинномозговых и, возможно, стволовых сенсомоторных механизмов. Результатом является ненормальное совместное сокращение мышц и перенапряжение мышц, прилегающих к основным движущим силам, или к противоположной части тела, так называемая зеркальная дистония. 14, 15
Существует большое количество доказательств того, что дисфункция нигростриатальной дофаминовой системы является патофизиологией дистонии.Хотя большая часть доказательств носит косвенный характер, нарушение нейротрансмиссии дофамина, по-видимому, является общей темой: (1) Лекарства, блокирующие дофаминовые рецепторы, могут вызывать острые дистонические реакции. 16 (2) При синдроме допа-чувствительной дистонии (DRD) наблюдается выраженная симптоматическая реакция на леводопу. 17 При DRD снижается продукция дофамина, несмотря на отсутствие потери нигростриатных нейронов. (3) У пациентов с идиопатической очаговой дистонией снижено связывание рецептора дофамина D 2 в скорлупе. 18 (4) Дистония является ранним проявлением болезни Паркинсона у 20-40% пациентов. 19 (5) Экспрессия матричной рибонуклеиновой кислоты (мРНК), кодирующей torsinA, который является белковым продуктом гена (DYT1), ответственного за идиопатическую генерализованную торсионную дистонию с началом в детстве, особенно сконцентрирована в нигростриатных дофаминовых нейронах. 20 (6) Дистония, связанная со «спастическим церебральным параличом», может поддаваться лечению леводопой, предшественником дофамина. 21 Хотя это не означает, что существует дефицит дофамина при спастическом церебральном параличе, это подтверждает мнение о том, что механизмы дофамина могут играть важную роль в патофизиологии дистонии.
Большинство исследований дистонии выявили аномальное совместное сокращение мышц-агонистов и антагонистов в состоянии покоя, которое часто усугубляется движением 14, 22–24 ; однако одно исследование с участием детей с первичной и вторичной дистонией не подтвердило одновременное сокращение как основание для дистонии. 25 В состоянии покоя ненормальные сокращения могут быть продолжительными или прерывистыми. Прерывистые сокращения обычно длятся от 1 до 2 секунд, хотя некоторые из них достигают 100 мс. Во время произвольного движения наблюдаются чрезмерные произвольные сокращения в мышцах, которые обычно не были бы активны при выполнении задания. В дополнение к чрезмерной активности близлежащих мышц, активность мышц-агонистов увеличивается, 26 и связанные с движением корковые потенциалы снижаются непосредственно перед движением. 27 Сочетание небольшого снижения активности агонистов и чрезмерной активности мышц-антагонистов приводит к замедлению движений.Последовательности движений относительно более нарушены, чем отдельные компоненты. 28
При дистонии моносинаптические рефлексы и рефлексы растяжения с длительным латентным периодом являются нормальными при высоких темпах растяжения, но при более медленных растяжениях рефлекс с длительным латентным периодом удлиняется по сравнению с нормальным. 29 Растяжение одной группы мышц часто приводит к активации «переполнения» других групп мышц, 24 и было высказано предположение, что люди с дистонией нарушают реципрокное торможение на супраспинальной основе. 30, 31 У людей с дистонией часто возникает парадоксальная активация пассивно укороченной мышцы (так называемая реакция укорочения). 24 Таким образом, различные механизмы могут способствовать аномальной мышечной активности, наблюдаемой при дистонии. Что общего у этих механизмов, так это неспособность подавлять нежелательную рефлекторную активность в ответ на растяжку. 11 Эта точка зрения подтверждается данными о том, что очаговая дистония может быть уменьшена путем блокирования движения γ-мотонейронов к веретенам с помощью лидокаина. 32
Клинический спектр допа-зависимой дистонии и связанных с ней заболеваний
Сегава М., Оми К., Ито С., Аояма М., Хаякава Х. Болезнь базальных ганглиев в детстве с выраженной реакцией на L-допа: наследственно прогрессирующая базальная заболевание ганглиев с выраженными суточными колебаниями. Шинрё. 1972; 24: 667–72.
Google Scholar
Beck D. Dystonia musculosum deformans с другим случаем из той же семьи.Proc R Soc Med. 1947; 40: 551–2.
CAS PubMed Central PubMed Google Scholar
Уголок BD. Dystonia musculorum deformans у братьев и сестер: лечение артаном (тригексифенидил). Proc R Soc Med. 1952, 45 (7): 451–2.
CAS PubMed Google Scholar
Раджпут AH. Леводопа при деформирующей мышечной дистонии. Ланцет. 1973; 1: 432.
CAS PubMed Статья Google Scholar
Ouvrier RA. Прогрессирующая дистония с выраженными суточными колебаниями. Энн Нейрол. 1978. 4 (5): 412–7. DOI: 10.1002 / ana.410040505.
CAS PubMed Статья Google Scholar
Гордон Н. Колеблющаяся дистония и родственные синдромы. Нейропедиатрия. 1982. 13 (3): 152–4. DOI: 10,1055 / с-2008-1059614.
CAS PubMed Статья Google Scholar
Kumammoto I, Nomoto M, Yoshodome M, Osame M, Igata A.Пять случаев дистонии с выраженными суточными колебаниями и особой ссылкой на гомованилиновую кислоту в спинномозговой жидкости. Clin Neurol (Токио). 1984; 24: 697–702.
Google Scholar
Sunohara N, Mano Y, Ando K, Satoyoshi E. Идиопатическая дистония-паркинсонизм с выраженными суточными колебаниями симптомов. Энн Нейрол. 1985. 17 (1): 39–45. DOI: 10.1002 / ana.410170110.
CAS PubMed Статья Google Scholar
Деонна Т. ДОПА-чувствительная прогрессирующая дистония детского возраста с колебаниями симптомов — синдром Сегавы и возможные варианты. Результаты совместного исследования Европейской федерации обществ детских неврологов (EFCNS). Нейропедиатрия. 1986. 17 (2): 81–5.
CAS PubMed Статья Google Scholar
Nygaard TG, Duvoisin RC. Наследственная дистония-паркинсонизм с юношеским началом. Неврология. 1986. 36 (11): 1424–8.
CAS PubMed Статья Google Scholar
de Yebenes JG, Moskowitz C, Fahn S, Saint-Hilaire MH. Длительное лечение леводопой в семье с аутосомно-доминантной торсионной дистонией. Adv Neurol. 1988; 50: 101–11.
PubMed Google Scholar
Nygaard TG, Marsden CD, Duvoisin RC. Допа-чувствительная дистония. Adv Neurol. 1988. 50: 377–84.
CAS PubMed Google Scholar
Kim JW, Cho GS, Myung HJ. Две семьи наследственной прогрессирующей дистонии с суточными колебаниями. J Korean Neurol Assoc. 1990; 8: 389–92.
Google Scholar
Nygaard TG, Trugman JM, de Yebenes JG, Fahn S. Допа-чувствительная дистония: спектр клинических проявлений в большой североамериканской семье. Неврология.1990; 40 (1): 66–9.
CAS PubMed Статья Google Scholar
Чон Б.С., Чон Дж. М., Пак С. С., Ким Дж. М., Чанг Ю.С., Сон Х.С. и др. Плотность переносчика дофамина, измеренная с помощью однофотонной эмиссионной компьютерной томографии [123I] beta-CIT, является нормальной при допа-чувствительной дистонии. Энн Нейрол. 1998. 43 (6): 792–800. DOI: 10.1002 / ana.410430614.
CAS PubMed Статья Google Scholar
Jeon BS, Jeong JM, Park SS, Lee MC. Допа-чувствительная дистония: синдром селективной нигростриатальной недостаточности дофамина. В: Fahn S, Marsden CD, DeLong M, редакторы. Дистония 3: достижения неврологии, т. 78. Филадельфия: Липпинкотт-Рэйвен; 1998. с. 309–17.
Google Scholar
Сегава М. Наследственная прогрессирующая дистония с выраженными суточными колебаниями. Brain Dev. 2000; 22 Приложение 1: S65–80.
PubMed Статья Google Scholar
Ким Х.Дж., Чон Б.С., Ян Х.Д., Чо Дж.Й. Мне нужно что-то получше, чем сон. Ланцет. 2013; 381 (9866): 598. DOI: 10.1016 / S0140-6736 (12) 61892-3. Эта статья показала, что значительное улучшение сна может иметь место при постэнцефалитическом паркинсонизме, а также при допа-зависимой дистонии .
PubMed Статья Google Scholar
Ямамура Ю., Хаттори Н., Мацумин Н., Кузухара С., Мизуно Ю. Аутосомно-рецессивный паркинсонизм с ранним началом и суточными колебаниями: клинико-патологические характеристики и молекулярно-генетическая идентификация.Brain Dev. 2000; 22 Приложение 1: S87–91.
PubMed Статья Google Scholar
Чанг С.Дж., Пак ХК, Ки С.С., Ким МДж, Ли М.К. Заметные суточные колебания и благоприятный отдых у пациента с мутацией Паркина. Mov Disord. 2008. 23 (4): 624–6. DOI: 10.1002 / mds.21951.
PubMed Статья Google Scholar
Найгаард Т.Г., Варан СП, Левин Р.А., Наини А.Б., Чуториан А.М.Допа-зависимая дистония, имитирующая церебральный паралич. Pediatr Neurol. 1994. 11 (3): 236–40.
CAS PubMed Статья Google Scholar
Nygaard TG, Takahashi H, Heiman GA, Snow BJ, Fahn S, Calne DB. Долгосрочный ответ на лечение и позитронно-эмиссионное томографическое сканирование флюородопы при паркинсонизме в семье с допа-зависимой дистонией. Энн Нейрол. 1992. 32 (5): 603–8. DOI: 10.1002 / ana.410320502.
CAS PubMed Статья Google Scholar
Чайла Э.С., Маккейб Диджей, Деланти Н, Костелло Диджей, Мерфи РП. Расширение фенотипа допа-зависимой дистонии с началом в детстве. Arch Neurol. 2006. 63 (8): 1185–8. DOI: 10.1001 / archneur.63.8.1185.
PubMed Статья Google Scholar
Фурукава Ю., Киш С.Дж., Бебин Е.М., Якобсон Р.Д., Фрайбург Дж.С., Уилсон В.Г. и др. Дистония с моторной задержкой у сложных гетерозигот по мутациям гена GTP-циклогидролазы I. Энн Нейрол. 1998. 44 (1): 10–6.DOI: 10.1002 / ana.410440107.
CAS PubMed Статья Google Scholar
Харвуд Г., Хиеронс Р., Флетчер Н.А., Марсден, компакт-диск. Уроки замечательной семьи с допа-зависимой дистонией. J Neurol Neurosurg Psychiatry. 1994. 57 (4): 460–3.
CAS PubMed Central PubMed Статья Google Scholar
Tassin J, Durr A, Bonnet AM, Gil R, Vidailhet M, Lucking CB и др.Леводопа-зависимая дистония. ГТФ-циклогидролаза I или мутации паркина? Мозг. 2000; 123: 1112–21.
PubMed Статья Google Scholar
Сегава М., Номура Ю., Нишияма Н. Аутосомно-доминантный дефицит гуанозинтрифосфатциклогидролазы I (болезнь Сегавы). Энн Нейрол. 2003; 54 Приложение 6: S32–45. DOI: 10.1002 / ana.10630.
CAS PubMed Статья Google Scholar
Furukawa Y, Mizuno Y, Narabayashi H. Ранний паркинсонизм с дистонией. Клинические и биохимические отличия от наследственной прогрессирующей дистонии или DOPA-зависимой дистонии. Adv Neurol. 1996; 69: 327–37.
CAS PubMed Google Scholar
Фурукава Ю., Симадзу М., Раджпут А.Х., Симидзу Ю., Тагава Т., Мори Н. и др. Мутации гена GTP-циклогидролазы I при наследственной прогрессирующей и допа-зависимой дистонии. Энн Нейрол.1996. 39 (5): 609–17. DOI: 10.1002 / ana.4103.
CAS PubMed Статья Google Scholar
Блау Н., Бонафе Л., Тони Б. Дефицит тетрагидробиоптерина без гиперфенилаланинемии: диагностика и генетика допа-зависимой дистонии и дефицита сепиаптеринредуктазы. Mol Genet Metab. 2001. 74 (1–2): 172–85. DOI: 10.1006 / mgme.2001.3213.
CAS PubMed Статья Google Scholar
Хайланд К., Гунасекара Р.С., Мунк-Мартин Т.Л., Арнольд Л.А., Энгл Т. Мышь hph-1: модель доминантно наследуемой недостаточности GTP-циклогидролазы. Энн Нейрол. 2003; 54 Приложение 6: S46–8. DOI: 10.1002 / ana.10695.
CAS PubMed Статья Google Scholar
Фурукава Ю., Капатос Дж., Хейкок Дж. У., Уорсли Дж., Вонг Х., Киш С. Дж. И др. Биоптерин головного мозга и тирозингидроксилаза при бессимптомной допа-чувствительной дистонии. Энн Нейрол.2002. 51 (5): 637–41. DOI: 10.1002 / ana.10175.
CAS PubMed Статья Google Scholar
Rajput AH, Gibb WR, Zhong XH, Shannak KS, Kish S, Chang LG и др. Допа-зависимая дистония: патолого-биохимические наблюдения в кейсе. Энн Нейрол. 1994. 35 (4): 396–402. DOI: 10.1002 / ana.410350405.
CAS PubMed Статья Google Scholar
Sawle GV, Leenders KL, Brooks DJ, Harwood G, Lees AJ, Frackowiak RS и др. Допа-чувствительная дистония: [18F] допа-позитронно-эмиссионная томография. Энн Нейрол. 1991. 30 (1): 24–30. DOI: 10.1002 / ana.410300106.
CAS PubMed Статья Google Scholar
Окада А., Накамура К., Сноу Б.Дж., Бхатт М.Х., Номото М., Осаме М. и др. Исследование с помощью ПЭТ дофаминергической системы у японского пациента с наследственной прогрессирующей дистонией (болезнь Сегавы).В: Нагацу Т., Янагисава Н., Мидзуно Ю., редакторы. Успехи неврологии, т. 60. Нью-Йорк: Рэйвен; 1993. стр. 591–4.
Google Scholar
Snow BJ, Nygaard TG, Takahashi H, Calne DB. Позитронно-эмиссионные томографические исследования допа-зависимой дистонии и идиопатического паркинсонизма с ранним началом. Энн Нейрол. 1993. 34 (5): 733–8. DOI: 10.1002 / ana.410340518.
CAS PubMed Статья Google Scholar
Тони Б., Блау Н. Мутации в генах GTP-циклогидролазы I и 6-пирувоил-тетрагидроптеринсинтазы. Hum Mutat. 1997. 10 (1): 11–20. DOI: 10.1002 / (SICI) 1098-1004 (1997) 10: 1 <11 :: AID-HUMU2> 3.0.CO; 2-P.
CAS PubMed Статья Google Scholar
Хайланд К., Найгаард Т.Г., Тругман Дж.М., Свобода К.Дж., Арнольд Л.А., Спарагана С.П. Профили пероральной нагрузки фенилаланином у носителей гена с симптомами и бессимптомно с допа-чувствительной дистонией из-за доминантно наследуемой недостаточности циклогидролазы GTP.J Inherit Metab Dis. 1999. 22 (3): 213–5.
CAS PubMed Статья Google Scholar
Сараива Дж. М., Сикинс Дж. В., Смит И. Уровни фенилаланина и тирозина в плазме крови повторно проверены у гетерозигот на предмет гиперфенилаланинемии. J Inherit Metab Dis. 1993. 16 (1): 105–9.
CAS PubMed Статья Google Scholar
Saunders-Pullman R, Blau N, Hyland K, Zschocke J, Nygaard T., Raymond D, et al.Нагрузка фенилаланином как диагностический тест для DRD: интерпретация полезности теста. Mol Genet Metab. 2004. 83 (3): 207–12. DOI: 10.1016 / j.ymgme.2004.07.010.
CAS PubMed Статья Google Scholar
Furukawa Y, Nishi K, Kondo T., Mizuno Y, Narabayashi H. Уровни биоптерина в спинномозговой жидкости и клинические особенности пациентов с ювенильным паркинсонизмом. В: Нагацу Т., Янагисава Н., Мидзуно Ю., редакторы. Успехи неврологии, т.60, т. 60. Нью-Йорк: Рэйвен; 1993. стр. 562–7.
Google Scholar
Такахаши Х., Левин Р.А., Галлоуэй М.П., Сноу Б.Дж., Калне ДБ, Найгаард Т.Г. Биохимические данные и результаты позитронно-эмиссионной томографии фтородопа у бессимптомного носителя гена допа-чувствительной дистонии. Энн Нейрол. 1994. 35 (3): 354–6. DOI: 10.1002 / ana.410350317.
CAS PubMed Статья Google Scholar
Blau N, Joller P, Atares M, Cardesa-Garcia J, Niederwieser A. Повышение активности GTP-циклогидролазы I в мононуклеарных клетках крови путем стимуляции: обнаружение гетерозигот дефицита GTP-циклогидролазы I. Clin Chim Acta. 1985. 148 (1): 47–52.
CAS PubMed Статья Google Scholar
Ichinose H, Ohye T, Takahashi E, Seki N, Hori T., Segawa M, et al. Наследственная прогрессирующая дистония с выраженными суточными колебаниями, вызванная мутациями в гене циклогидролазы I GTP.Нат Жене. 1994. 8 (3): 236–42. DOI: 10,1038 / NG1194-236.
CAS PubMed Статья Google Scholar
Бонафе Л., Тони Б., Леймбахер В., Кират Л., Блау Н. Диагностика допа-зависимой дистонии и других нарушений тетрагидробиоптерина путем изучения метаболизма биоптерина в фибробластах. Clin Chem. 2001. 47 (3): 477–85.
CAS PubMed Google Scholar
Hirano M, Tamaru Y, Nagai Y, Ito H, Imai T., Ueno S. Пропуск экзона, вызванный заменой основания в сайте сплайсинга в гене циклогидролазы I GTP в японской семье с наследственной прогрессирующей дистонией, зависимой от допа дистонии. Biochem Biophys Res Commun. 1995. 213 (2): 645–51. DOI: 10.1006 / bbrc.1995.2180.
CAS PubMed Статья Google Scholar
Innis RB, Seibyl JP, Scanley BE, Laruelle M, Abi-Darham A, Wallace E, et al.Однофотонная эмиссионная компьютерная томография демонстрирует потерю переносчиков дофамина в полосатом теле при болезни Паркинсона. Proc Natl Acad Sci U S. A. 1993; 90 (24): 11965–9.
CAS PubMed Central PubMed Статья Google Scholar
Frost JJ, Rosier AJ, Reich SG, Smith JS, Ehlers MD, Snyder SH, et al. Позитронно-эмиссионная томография транспортера дофамина с помощью 11C-WIN 35,428 выявила заметное снижение заболеваемости болезнью Паркинсона в легкой форме.Энн Нейрол. 1993. 34 (3): 423–31. DOI: 10.1002 / ana.410340331.
CAS PubMed Статья Google Scholar
Marek KL, Seibyl JP, Zoghbi SS, Zea-Ponce Y, Baldwin RM, Fussell B, et al. Визуализация [123I] beta-CIT / SPECT демонстрирует двустороннюю потерю переносчиков дофамина при болезни геми-Паркинсона. Неврология. 1996. 46 (1): 231–7.
CAS PubMed Статья Google Scholar
Бандманн О., Найгаард Т.Г., Сёртиз Р., Марсден К.Д., Вуд Н.З., Хардинг А.Е. Допа-чувствительная дистония у британских пациентов: новые мутации гена GTP-циклогидролазы I и доказательства генетической гетерогенности. Hum Mol Genet. 1996. 5 (3): 403–6.
CAS PubMed Статья Google Scholar
Furukawa Y, Lang AE, Trugman JM, Bird TD, Hunter A, Sadeh M, et al. Гендерная пенетрантность и мутации de novo гена GTP-циклогидролазы I при допа-чувствительной дистонии.Неврология. 1998. 50 (4): 1015–20.
CAS PubMed Статья Google Scholar
Suzuki T, Ohye T, Inagaki H, Nagatsu T, Ichinose H. Характеристика дикого типа и мутантов рекомбинантной GTP циклогидролазы I человека: связь с этиологией допа-зависимой дистонии. J Neurochem. 1999. 73 (6): 2510–6.
CAS PubMed Статья Google Scholar
Hagenah J, Saunders-Pullman R, Hedrich K, Kabakci K, Habermann K, Wiegers K и др. Высокая частота мутаций при допа-чувствительной дистонии: обнаружение с помощью комплексного скрининга GCHI. Неврология. 2005. 64 (5): 908–11. DOI: 10.1212 / 01.WNL.0000152839.50258.A2.
CAS PubMed Статья Google Scholar
Zirn B, Steinberger D, Troidl C, Brockmann K, von der Hagen M, Feiner C, et al. Частота делеций GCh2 при допа-чувствительной дистонии.J Neurol Neurosurg Psychiatry. 2008. 79 (2): 183–6. DOI: 10.1136 / jnnp.2007.128413.
CAS PubMed Статья Google Scholar
Hahn H, Trant MR, Brownstein MJ, Harper RA, Milstien S, Butler IJ. Неврологические и психиатрические проявления в семье с мутацией во 2 экзоне гена гуанозинтрифосфатциклогидролазы. Arch Neurol. 2001. 58 (5): 749–55.
CAS PubMed Статья Google Scholar
Мори С., Мацуяма К., Кохьяма Дж., Кобаяши Ю., Такакусаки К. Нейронные составляющие систем постурального и локомоторного контроля и их взаимодействия у кошек. Brain Dev. 1992; 14 (Дополнение): S109–20.
PubMed Google Scholar
Левитт П., Харви Дж. А., Фридман Е., Симански К., Мерфи Е. Х. Новые данные о влиянии нейротрансмиттеров на развитие мозга. Trends Neurosci. 1997. 20 (6): 269–74.
CAS PubMed Статья Google Scholar
Комори Х., Мацуиси Т., Ямада С., Уэда Н., Ямасита Ю., Като Х. Влияние возраста на уровни метаболитов биоптерина и биогенных аминов в спинномозговой жидкости. Acta Paediatr. 1999. 88 (12): 1344–7.
CAS PubMed Статья Google Scholar
Hwu WL, Wang PJ, Hsiao KJ, Wang TR, Chiou YW, Lee YM. Допа-чувствительная дистония, вызванная рецессивной мутацией циклогидролазы I GTP. Hum Genet. 1999. 105 (3): 226–30.
CAS PubMed Статья Google Scholar
Jarman PR, Bandmann O, Marsden CD, Wood NW. Мутации GTP циклогидролазы I у пациентов с дистонией, чувствительных к антихолинергическим препаратам. J Neurol Neurosurg Psychiatry. 1997. 63 (3): 304–8.
CAS PubMed Central PubMed Статья Google Scholar
Йунг В.Л., Вонг В.К., Чан К.Й., Хуэй Дж., Фунг К.В., Яу Э. и др. Расширение фенотипа и клинический анализ недостаточности тирозингидроксилазы. J Child Neurol. 2011; 26 (2): 179–87.DOI: 10.1177 / 0883073810377014.
PubMed Статья Google Scholar
Ribases M, Serrano M, Fernandez-Alvarez E, Pahisa S, Ormazabal A, Garcia-Cazorla A, et al. Гомозиготная мутация промотора гена тирозингидроксилазы у пациента с допа-чувствительной энцефалопатией: клинический, биохимический и генетический анализ. Mol Genet Metab. 2007. 92 (3): 274–7. DOI: 10.1016 / j.ymgme.2007.07.004.
CAS PubMed Статья Google Scholar
Hoffmann GF, Assmann B, Brautigam C, Dionisi-Vici C, Haussler M, de Klerk JB и др. Дефицит тирозингидроксилазы вызывает прогрессирующую энцефалопатию и допа-невосприимчивую дистонию. Энн Нейрол. 2003; 54 Приложение 6: S56–65. DOI: 10.1002 / ana.10632.
CAS PubMed Статья Google Scholar
Людеке Б., Кнаппског П.М., Клейтон П.Т., Сёртиз Р.А., Клелланд Д.Д., Хилес С.Дж. и др. Рецессивно унаследованный L-ДОФА-чувствительный паркинсонизм в младенчестве, вызванный точечной мутацией (L205P) в гене тирозингидроксилазы.Hum Mol Genet. 1996. 5 (7): 1023–8.
CAS PubMed Статья Google Scholar
Schiller A, Wevers RA, Steenbergen GC, Blau N, Jung HH. Длительное течение L-допа-чувствительной дистонии, вызванной дефицитом тирозингидроксилазы. Неврология. 2004. 63 (8): 1524–6.
CAS PubMed Статья Google Scholar
Людеке Б., Дворничак Б., Бартоломе К.Точечная мутация в гене тирозингидроксилазы, связанная с синдромом Сегавы. Hum Genet. 1995. 95 (1): 123–5.
CAS PubMed Статья Google Scholar
Swaans RJ, Rondot P, Renier WO, Van Den Heuvel LP, Steenbergen-Spanjers GC, Wevers RA. Четыре новые мутации в гене тирозингидроксилазы у пациентов с детским паркинсонизмом. Энн Хам Жене. 2000. 64: 25–31. DOI: 10.1017 / S0003480000007922.
CAS PubMed Статья Google Scholar
Bonafe L, Thony B, Penzien JM, Czarnecki B, Blau N. Мутации в гене сепиаптеринредуктазы вызывают новый тетрагидробиоптерин-зависимый дефицит моноамин-нейромедиатора без гиперфенилаланинемии. Am J Hum Genet. 2001. 69 (2): 269–77. DOI: 10,1086 / 321970.
CAS PubMed Central PubMed Статья Google Scholar
Steinberger D, Blau N, Goriuonov D, Bitsch J, Zuker M, Hummel S, et al. Гетерозиготная мутация в 5′-нетранслируемой области гена сепиаптеринредуктазы (SPR) у пациента с допа-чувствительной дистонией.Нейрогенетика. 2004. 5 (3): 187–90. DOI: 10.1007 / s10048-004-0182-3.
CAS PubMed Статья Google Scholar
Невилл Б.Г., Параскандало Р., Фарруджа Р., Феличе А. Дефицит редуктазы сепиаптерина: врожденное допа-зависимое моторное и когнитивное расстройство. Мозг. 2005; 128: 2291–6. DOI: 10,1093 / мозг / awh603.
CAS PubMed Статья Google Scholar
Abeling NG, Duran M, Bakker HD, Stroomer L, Thony B, Blau N и др. Дефицит сепиаптеринредуктазы — аутосомно-рецессивная ДОФА-зависимая дистония. Mol Genet Metab. 2006. 89 (1–2): 116–20. DOI: 10.1016 / j.ymgme.2006.03.010.
CAS PubMed Статья Google Scholar
Эхенн Б., Руберти А., Ассманн Б., Лутц Т., Пензиен Дж. М., Тони Б. и др. Дефицит сепиаптеринредуктазы: клинические проявления и оценка длительной терапии.Pediatr Neurol. 2006. 35 (5): 308–13. DOI: 10.1016 / j.pediatrneurol.2006.05.006.
PubMed Статья Google Scholar
Фридман Дж., Хайланд К., Блау Н., Макколлин М. Допа-чувствительная гиперсомния и смешанное двигательное расстройство из-за недостаточности сепиаптеринредуктазы. Неврология. 2006. 67 (11): 2032–205. DOI: 10.1212 / 01.wnl.0000247274.21261.b4.
PubMed Статья Google Scholar
Аррабаль Л., Тереза Л., Санчес-Алькудия Р., Кастро М., Медрано С., Гутьеррес-Солана Л. и др. Взаимосвязь генотипа и фенотипа при дефиците сепиаптеринредуктазы. Дефект сплайсинга составляет новый фенотипический вариант. Нейрогенетика. 2011; 12 (3): 183–91. DOI: 10.1007 / s10048-011-0279-4.
CAS PubMed Статья Google Scholar
Фридман Дж., Розе Э., Абденур Дж. Э., Чанг Р., Гасперини С., Салетти В. и др. Дефицит сепиаптеринредуктазы: излечимая имитация церебрального паралича.Энн Нейрол. 2012. 71 (4): 520–30. DOI: 10.1002 / ana.22685.
CAS PubMed Статья Google Scholar
Куриан М.А., Ли Й., Чжэнь Дж., Мейер Э., Хай Н., Кристен Х. Дж. И др. Клиническая и молекулярная характеристика синдрома наследственной недостаточности переносчика дофамина: когортное наблюдение и экспериментальное исследование. Lancet Neurol. 2011; 10 (1): 54–62. DOI: 10.1016 / S1474-4422 (10) 70269-6. В этом отчете описывается недавно выявленная транспортопатия с мутацией в гене переносчика дофамина (SLC6A3).У детей с дефицитом переносчика дофамина симптомы были очень похожи на симптомы пациентов с DRD-plus. Мы должны учитывать транспортопатии при ведении пациентов с манифестацией DRD-plus .
CAS PubMed Статья Google Scholar
Рилстон Дж. Дж., Алхатер Р.А., Минасян Б.А. Болезнь везикулярного транспорта дофамина-серотонина в мозге и ее лечение. N Engl J Med. 2013. 368 (6): 543–50. DOI: 10.1056 / NEJMoa1207281. Rilstone, et al. сообщили о семье с мутацией в SLC18A2, который кодирует везикулярный транспортер моноаминов 2. У пораженных членов наблюдались симптомы, связанные с дисфункцией дофамина, серотонина и адреналина / норэпинефрина. Это еще одна транспортопатия, проявляющаяся как DRD-plus. Мы должны учитывать транспортопатии при ведении пациентов с манифестацией DRD-plus .
CAS PubMed Статья Google Scholar
Van Hove JL, Steyaert J, Matthijs G, Legius E, Theys P, Wevers R, et al. Расширенный моторный и психиатрический фенотип при аутосомно-доминантном синдроме Сегавы из-за дефицита циклогидролазы GTP. J Neurol Neurosurg Psychiatry. 2006. 77 (1): 18–23. DOI: 10.1136 / jnnp.2004.051664.
PubMed Central PubMed Статья Google Scholar
Yaltho TC, Jankovic J, Lotze T. Связь синдрома Туретта и допа-зависимой дистонии.Mov Disord. 2011; 26 (2): 359–60. DOI: 10.1002 / mds.23424.
PubMed Статья Google Scholar
Bandmann O, Valente EM, Holmans P, Surtees RA, Walters JH, Wevers RA, et al. Допа-чувствительная дистония: клиническое и молекулярно-генетическое исследование. Энн Нейрол. 1998. 44 (4): 649–56. DOI: 10.1002 / ana.410440411.
CAS PubMed Статья Google Scholar
Regula JU, Thoden U, Meinck HM. Дистония у взрослых: атипичное проявление болезни Сегавы. Mov Disord. 2007. 22 (9): 1335–7. DOI: 10.1002 / mds.21377.
PubMed Статья Google Scholar
Ли Дж.Й., Ян Х.Дж., Ким Дж.М., Чон Б.С. Новые мутации GCH-1 и необычные длительные дискинезии в корейских семьях с допа-зависимой дистонией. Паркинсонизм, связанный с разладом. 2013; 19 (12): 1156–9. DOI: 10.1016 / j.parkreldis.2013.08.003. Это отчет о корейских случаях DRD с классическими проявлениями. У всех пациентов были обнаружены мутации GCH-1. Следует отметить, что в первоначальном сообщении (ссылка 15) не удалось показать мутацию в GCH-1 в некоторых из них, в то время как они предсказывали наличие мутации в GCH-1 из-за низкого уровня неоптерина в спинномозговой жидкости.
PubMed Статья Google Scholar
Тадич В., Кастен М., Брюггеманн Н., Стиллер С., Хагена Дж., Кляйн К.Пересмотр допа-зависимой дистонии: задержка диагностики, остаточные и немоторные признаки. Arch Neurol. 2012: 1–5. DOI: 10.1001 / archneurol.2012.574. Авторы сообщили о высокой частоте остаточных моторных признаков у пролеченных пациентов с DRD, что несовместимо с нашим опытом (ссылка 82) и нашим прогнозом, что L-допа должна полностью изменить моторные симптомы (кроме контрактур).
Van Gerpen JA. Допа-чувствительная дистоническая камптокормия.Неврология. 2006; 66 (11): 1779. DOI: 10.1212 / 01.wnl.0000218158.61678.55.
PubMed Статья Google Scholar
Schneider SA, Mohire MD, Trender-Gerhard I., Asmus F, Sweeney M, Davis M, et al. Семейная допа-зависимая шейная дистония. Неврология. 2006. 66 (4): 599–601. DOI: 10.1212 / 01.wnl.0000198501.61063.66.
CAS PubMed Статья Google Scholar
Charlesworth G, Mohire MD, Schneider SA, Stamelou M, Wood NW, Bhatia KP. Атаксия, телеангиэктазия, проявляющаяся как допа-зависимая цервикальная дистония. Неврология. 2013. 81 (13): 1148–51. DOI: 10.1212 / WNL.0b013e3182a55fa2. Это очень своеобразное семейство с допа-чувствительной цервикальной дистонией и мутацией в гене атаксии-телеангиэктазии. Связь между реакцией на L-допа и мутацией при атаксии-телеангиэктазии сомнительна, учитывая ссылку 87, где дистония не реагировала на L-допа у испытуемых пациентов .
PubMed Central PubMed Статья Google Scholar
Saunders-Pullman R, Raymond D, Stoessl AJ, Hobson D, Nakamura K, Pullman S, et al. Вариант атаксии-телеангиэктазии, проявляющейся первичной дистонией у канадских меннонитов. Неврология. 2012. 78 (9): 649–57. DOI: 10.1212 / WNL.0b013e3182494d51.
CAS PubMed Central PubMed Статья Google Scholar
Ichinose H, Inagaki H, Suzuki T, Ohye T., Nagatsu T. Молекулярные механизмы наследственной прогрессирующей дистонии с заметными суточными колебаниями. Болезнь Сегавы. Brain Dev. 2000; 22 Приложение 1: S107–10.
PubMed Статья Google Scholar
Уидер С., Мелквист С., Хауф М., Солида А., Кобб С.А., Кахергус Дж. М. и др. Исследование швейцарской семьи допа-чувствительных дистоний с делецией в GCh2: переопределение DYT14 как DYT5. Неврология.2008. 70 (16): 1377–83. DOI: 10.1212 / 01.wnl.0000275527.35752.c5.
CAS PubMed Central PubMed Статья Google Scholar
Нагата Е., Косакай А., Танака К., Сегава М., Фудзиока Х., Синтаку Х. и др. Допа-чувствительная дистония (болезнь Сегавы) -подобное заболевание, сопровождающееся умственной отсталостью: клинический случай. Mov Disord. 2007. 22 (8): 1202–3. DOI: 10.1002 / mds.21517.
PubMed Статья Google Scholar
Hjermind LE, Johannsen LG, Blau N, Wevers RA, Lucking CB, Hertz JM, et al. Допа-чувствительная дистония и болезнь Паркинсона с ранним началом у пациента с дефицитом циклогидролазы I GTP? Mov Disord. 2006. 21 (5): 679–82. DOI: 10.1002 / mds.20773.
PubMed Статья Google Scholar
Кикучи А., Такеда А., Фуджихара К., Кимпара Т., Шига Ю., Танджи Х. и др. Arg (184) Его мутантная GTP циклогидролаза I, вызывающая рецессивную гиперфенилаланинемию, ответственна за допа-чувствительную дистонию с паркинсонизмом: отчет о клиническом случае.Mov Disord. 2004. 19 (5): 590–3. DOI: 10.1002 / mds.10712.
PubMed Статья Google Scholar
Ichinose H, Ohye T, Matsuda Y, Hori T, Blau N, Burlina A, et al. Характеристика генов GTP-циклогидролазы I мыши и человека. Мутации у пациентов с дефицитом ГТФ-циклогидролазы I. J Biol Chem. 1995. 270 (17): 10062–71.
CAS PubMed Статья Google Scholar
Эггерс С., Фольк А.Е., Кахраман Д., Финк Г.Р., Лейбе Б., Шмидт М. и др. Связаны ли допа-зависимая дистония и болезнь Паркинсона? Отчет о болезни. Паркинсонизм, связанный с разладом. 2012; 18 (5): 666–8. DOI: 10.1016 / j.parkreldis.2011.10.003.
PubMed Статья Google Scholar
Найгаард Т.Г. Допа-чувствительная дистония: описание клинического синдрома и ключи к патогенезу. В: Нагацу Т., Янагисава Н., Мидзуно Ю., редакторы.Успехи неврологии, т. 60. Нью-Йорк: Рэйвен; 1993. стр. 577–85.
Google Scholar
Nygaard TG, Marsden CD, Fahn S. Допа-чувствительная дистония: долгосрочный ответ на лечение и прогноз. Неврология. 1991. 41 (2): 174–81.
CAS PubMed Статья Google Scholar
Turjanski N, Bhatia K, Burn DJ, Sawle GV, Marsden CD, Brooks DJ. Сравнение поглощения 18F-допа полосатым телом при дистонии-паркинсонизме с началом у взрослых, болезни Паркинсона и допа-зависимой дистонии.Неврология. 1993. 43 (8): 1563–8.
CAS PubMed Статья Google Scholar
Jellinger K. Паркинсонизм с юношеским началом: один и тот же пациент сообщил дважды. Неврология. 1992. 42 (5): 1124–5.
CAS PubMed Статья Google Scholar
Clot F, Grabli D, Cazeneuve C, Roze E, Castelnau P, Chabrol B, et al. Исчерпывающий анализ генов биосинтеза Bh5 и дофамина у пациентов с допа-зависимой дистонией.Мозг. 2009; 132: 1753–63. DOI: 10,1093 / мозг / awp084.
PubMed Статья Google Scholar
Диагностика и лечение дистонии | Центр головного мозга и позвоночника Weill Cornell
Врач изучит всю историю болезни и проведет медицинский осмотр для выявления признаков и симптомов заболевания. Могут быть заказаны другие тесты с целью определения места возникновения проблемы и исключения других условий. Эти тесты включают:
Анализ крови или мочи: Эти тесты выявят токсины в организме
Электроэнцефалограммы (ЭЭГ): Этот тест регистрирует некоторые аспекты электрической активности мозга.Это может выявить другие основные проблемы, которые могут помочь определить причину дистонии.
Компьютерная томография (КТ) — это неинвазивная процедура, при которой для получения трехмерного изображения мозга используются рентгеновские лучи. Мозг с дистонией выглядит нормальным при компьютерной томографии; однако сканирование может выявить и другие условия.
Магнитно-резонансная томография (МРТ) использует магнитные поля и радиоволны для создания детального изображения мозга. Этот тест можно использовать для выявления других состояний, таких как инсульт или опухоли головного мозга.
Электромиография (ЭМГ): Этот тест измеряет электрическую активность в мышцах.
Эти диагностические тесты могут помочь врачу определить состояние, тем самым обеспечивая правильный диагноз и лечение.
Варианты лечения
Хотя от дистонии нет лекарства, существуют методы лечения, которые значительно улучшают симптомы.
Лекарства: Существуют пероральные и инъекционные лекарства, которые могут помочь при мышечных спазмах и сокращениях.Некоторые из этих лекарств, в том числе баклофен, работают за счет увеличения количества дофамина — нейромедиатора, участвующего в движении мышц — в мозге. Инъекции ботулинического нейротоксина (ботокса) расслабляют мышцы и облегчают сокращения
Therapy: Регулярная физиотерапия и логопедия могут помочь пациенту справиться с симптомами дистонии и улучшить их.
Хирургия: глубокая стимуляция мозга (DBS) — это хирургическая процедура для лечения неврологических симптомов, включая симптомы дистонии, включая тремор и контроль движений.Открытая операция иногда используется при дистонии и может быть вариантом для тех, кто не получил успешного лечения с помощью других методов лечения.
Baclofen Pumps: Это лечение используется при очаговой дистонии. Баклофен — это лекарство, которое облегчает движение мышц, и оно доставляется непрерывно через насос, имплантированный в брюшную полость. Двумя типами доставки являются интратекальный (с доставкой лекарства в спинномозговую жидкость) и внутрижелудочковый (с доставкой лекарства в желудочки мозга).
Для получения дополнительной информации см. Хирургия дистонии.
Узнайте больше об услугах по лечению двигательных расстройств и программе по педиатрическим нейромоторным расстройствам в Центре мозга и позвоночника Weill Cornell или воспользуйтесь нашей онлайн-формой, чтобы записаться на прием.